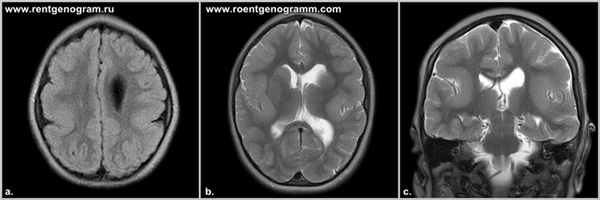
Лиссэнцефалия - агирия. Шизэнцефалия. Порэнцефалия. Голопрозэнцефалия
Обновлено: 04.05.2024
Пахигирия (от Греческий «пахи», что означает «толстые» или «толстые» извилины) является врожденным пороком развития полушария головного мозга. Это приводит к необычно толстым извилинам кора головного мозга. Обычно у детей отставание в развитии и припадки, начало и тяжесть в зависимости от тяжести коркового порока. Инфантильные спазмы часто встречаются у пораженных детей, поскольку трудноизлечимы эпилепсия.
Содержание
Презентация
Термин «пахигирия» не имеет прямого отношения к конкретному уродству, а скорее используется для общего описания физических характеристик мозга в сочетании с несколькими. нарушения миграции нейронов; чаще всего расстройства, относящиеся к разной степени лиссэнцефалия. Лиссэнцефалия присутствует у 1 из 85 470 рождений, и продолжительность жизни пострадавших невелика, так как лишь немногие доживают до 20 лет. [1] Пахигирия - это состояние, определяемое типом генетической аномалии коры головного мозга. Клиницисты будут субъективно определять уродство на основании степени дефекта и степени присутствующей утолщенной аномальной серой дифференцировки. [2]
Связь с эпилепсией, лиссэнцефалией и гетеротопией подкорковых полос
Различные степени выраженности и локализации эпилепсии связаны с пороками коркового развития. Исследователи предполагают, что примерно 40% детей с диагнозом лекарственно-устойчивая эпилепсия имеют некоторую степень коркового порока развития. [1] [2]
Лиссэнцефалия (с которой наиболее тесно связана пахигирия) связана с тяжелым умственная отсталость, эпилепсия, и нарушение моторики. Две характеристики лиссэнцефалии включают отсутствие извилины (агирия ) и уменьшение наличия извилин (пахигирия). [2] Типы судорог, связанных с лиссэнцефалией, включают:
- стойкие спазмы
- фокальные припадки
- тонические припадки
- атипичные припадки
- атонические припадки [1]
Другие возможные симптомы лиссэнцефалии включают: телекантус, эстропия, гипертелоризм, разная степень умственной отсталости, мозжечок гипоплазия, мозолистое тело аплазия, и уменьшился мышечный тонус и сухожильные рефлексы. [3] Более 90% детей с лиссэнцефалией страдают судорогами. [2]
Пациенты с подкорковая полоса гетеротопия (другое расстройство, связанное с пахигирией) обычно имеют более легкие симптомы, и их когнитивная функция тесно связана с толщиной подкорковая полоса и степень присутствующей пахигирии. [2]
Причины
Пахигирия вызывается нарушением процесса миграции нейронов плода из-за генетических или, возможно, факторов окружающей среды. Пораженная кора головного мозга обычно имеет только четыре развитых слоя вместо обычных шести. Одним из наиболее известных и наиболее распространенных типов нарушений миграции нейронов является лиссэнцефалия диффузная корковая мальформация, непосредственно связанная с агирией и пахигирией. [4] Неполная миграция нейронов на раннем этапе развития мозга плода является предвестником лиссэнцефалии. [5] Если нейроны следуют аномальной миграции во время развития, возможные корковые аномалии включают: классическая лиссэнцефалия (как указано выше) и подкорковая полоса гетеротопия с полосой спектра агирии-пахигирии. [2]
Нормальная миграция нейронов
Нормальная миграция нейронов включает развитие шести корковых слоев, каждый из которых выполняет определенные функции. [2]
Нормальное мозговое развитие происходит в трех динамических и перекрывающихся стадиях:
- первая стадия: стволовые клетки пролиферируют и дифференцируются в нейроны или глиальные клетки в пределах передний мозг и желудочковые и субвентрикулярные зоны, выстилающие полость мозга;
- у людей эта стадия длится от 5-6 до 16-20 недель гестации;
- второй этап: миграция от начала координат по радиусу вдоль глиальные волокна от перивентрикулярная область ганглиозных возвышенностей к пиал поверхность;
- поколения укладываются в шаблон в корковая пластинка на этом этапе;
- у людей эта стадия длится от 6-7 до 20-24 недель гестации;
- у людей эта стадия длится от 16 недели гестации до долгого времени после рождения. [2]
Большинство типов неполной миграции нейронов в кору головного мозга происходит в течение третьего и четвертого месяцев беременности. [4] Аномальная миграция нейронов приводит к тому, что они не достигают своих конечных пунктов назначения, что приводит к отказу борозды и извилины для образования. [2]
Стадия коркового развития, на которой миграция прекращается, напрямую связана с уровнем структурного неправильного положения. [1]
Один из самых критических этапов в развитии мозга - это когда пост-митотический нейроны мигрируют из зоны желудочков с образованием корковой пластинки. [6] Миграция, остановленная на последней стадии развития, обычно ограничивает аномальное положение клеток уровнем коры. [1]
Нарушение миграции нейронов, вызванное генетическими мутациями
Было выделено несколько генетических мутаций, связанных с определенными пороками развития коры головного мозга. [1] Гены, вызывающие лизэнцефалию, включают как аутосомные, так и Х-сцепленные гены. [3] Ниже обсуждаются мутации генов LIS1 или DCX, поскольку они чаще всего связаны с нарушениями миграции нейронов, включая лиссэнцефалию-пахигирию и гетеротопию подкорковых полос. [2]
LIS1 отвечает за аутосомную форму лиссэнцефалии. [2] Мутации гена LIS1 связаны примерно с 80% больных лиссэнцефалией. [5] LIS1 был первым клонированным геном миграции нейронов человека. Он отвечает за кодирование альфа-субъединицы внутриклеточной изоформы Ib ацетилгидролазы фактора активации тромбоцитов. Он расположен на хромосоме 17p13.3 и имеет 11 экзонов с кодирующей областью 1233bp. Белок LIS1, по-видимому, взаимодействует с тубулином, подавляя динамику микротрубочек. Белок является высококонсервативным, и исследования показали, что он участвует в цитоплазматическом динеин-опосредованном нуклеокинезе, транслокации сомов, подвижности клеток, митозе и сегрегации хромосом. [6] LIS1 кодирует белок размером 45 кДа под названием PAFAH1B1, который содержит семь повторов WD40, необходимых для правильной миграции нейронов. [5] Ген LIS1 кодирует белок, похожий на β-субъединицу G-белков, ответственных за разложение биоактивного липидного фактора активации тромбоцитов (PAF). [2] Это приводит к теориям, что LIS1 может оказывать свое влияние на миграцию через микротрубочки. Конкретные концентрации PAF могут быть необходимы для оптимальной миграции нейронов, влияя на адгезионные свойства морфологии клеток. Исследования показали, что добавление PAF или ингибирование фактора активации тромбоцитов ацетилгидролазы (PAF-AH) снижает миграцию гранулярных клеток мозжечка. in vitro. Добавление PAF к клеткам гиппокампа показало коллапс конуса роста и ретракцию нейритов. Гомозиготные нулевые мыши с нокаутом LIS1 погибают во время эмбриогенеза, а гетерозиготные мыши выживают с отсроченной миграцией нейронов, подтвержденной in vitro и in vivo анализы миграции клеток. [5] Большинство случаев лиссэнцефалии связаны с делециями мутаций гена LIS1, и результаты обычно более тяжелые в задних областях мозга. [2]
Одно исследование показало, что из изолированной группы пациентов с лизэнцефалией 40% возникли в результате делеции LIS1, а еще 25% - в результате внутригенной мутации гена. Пациенты с миссенс-мутациями, как правило, имеют менее серьезные симптомы, пахигирию и редкие случаи гетеротопии подкорковых лент. Усеченные (укороченные) мутации LIS1 имеют тенденцию вызывать тяжелую лиссэнцефалию. [2]
Даблкортин
Даблкортин (DCX или XLIS) мутации ответственны за Х-сцепленные расстройства. [2] В то время как мутации LIS1 имеют тенденцию вызывать тяжелые уродства в задней части мозга, мутации DCX сосредотачивают большую часть своего разрушения на передних пороках и связаны с лиссэнцефалией у мужчин и гетеротопиями подкорковых лент у женщин. [2] [5] У женщин с мутациями DCX обычно преобладает передняя субкортикальная полостная гетеротопия и пахигирия. [1] [2] DCX был первым известным геном, вызывающим Х-сцепленную лизэнцефалию и гетеротопию подкорковых полос. Он обнаружен на хромосоме Xq22.3-q23 и имеет девять экзонов, которые кодируют 360 белков. DCX экспрессируется исключительно в мозге плода. [6]
Подробнее о наиболее распространенной форме церебрального паралича см. Спастическая диплегия.
Спастический церебральный паралич - наиболее распространенный тип, встречающийся в 70–80% всех случаев. Более того, спастический ХП в 30% случаев сопровождает один из других типов. Люди с этим типом страдают гипертонусом и имеют нервно-мышечное состояние, вызванное повреждением кортикоспинального тракта или моторной коры, которое влияет на способность нервной системы получать гамма-аминомасляную кислоту в областях, пораженных инвалидностью. Спастический ХП далее классифицируется по топографии в зависимости от пораженной области тела; они включают:
Спастическая гемиплегия (поражена одна сторона). Как правило, повреждение мышечных нервов, контролируемых левой стороной мозга, вызывает дефицит правого тела, и наоборот. Как правило, люди со спастической гемиплегией являются наиболее амбулаторными, хотя у них обычно имеется динамический эквинус на пораженной стороне, и им в первую очередь назначаются ортезы голеностопного сустава для предотвращения указанного эквинуса. [11] Спастическая диплегия (поражение нижних конечностей практически отсутствует. спастичность верхней части тела). Наиболее частая форма спастических форм. Большинство людей со спастической диплегией полностью амбулаторно и имеют походку «ножницы». Согнутые в той или иной степени колени и бедра - обычное явление. Проблемы с бедром, вывихи и у трех четвертей спастических диплегий также могут присутствовать косоглазие (косоглазие). Кроме того, эти люди часто близоруки. Это состояние не влияет на интеллект человека со спастической диплегией. Спастическая тетраплегия (поражаются все четыре конечности одинаково). У людей со спастической квадриплегией меньше всего шансов ходить или, если они могут, хотеть ходить, потому что их мышцы слишком напряжены, и для этого требуется слишком много усилий. У некоторых детей с квадриплегией также наблюдается гемипаретический тремор, неконтролируемое дрожание, которое поражает конечности с одной стороны тела и нарушает нормальное движение. Иногда такие термины, как моноплегия, параплегия, триплегия и пентаплегия, также могут использоваться для обозначения конкретных проявлений спастичность.
Патогенез
Пахигирия, лиссэнцефалия (гладкий мозг) и полимикрогирия (множественные маленькие извилины) - все это результат ненормального миграция клеток. Аномальная миграция обычно связана с неорганизованной клеточной архитектурой, неспособностью сформировать шесть слоев корковых нейронов (обычно четырехслойная кора) и функциональными проблемами. Аномальное образование головного мозга может быть связано с припадки, отставание в развитии, и психические расстройства.
В норме клетки мозга начинают развиваться в перивентрикулярная область (зародышевый матрикс ), а затем мигрируют от медиального к латеральному, чтобы сформировать кора головного мозга.
Диагностика
Для диагностики обычно используются различные методы визуализации. В то время как компьютерная томография (КТ) обеспечивает визуализацию головного мозга с более высоким пространственным разрешением, пороки развития коры головного мозга легче визуализируются in vivo и классифицированы с использованием магнитно-резонансная томография (МРТ ), что обеспечивает более контрастное изображение и лучшее очертание белый и серое вещество. [7]
Диффузную пахигирию (легкую форму лиссэнцефалии) можно увидеть на МРТ как утолщение коры головного мозга с небольшими и большими извилины и неполное развитие Сильвианские трещины. [3]
- серьезный эпилепсия[7]
- снижение продолжительности жизни
- разная степень умственной отсталости
- несговорчивый эпилепсия[5]
Познавательная способность коррелирует с толщиной любой имеющейся подкорковой полосы и степенью пахигирии. [1] [5]
Классификации
Степень порока развития коры головного мозга, вызванного: генетические мутации классифицируется по степени неправильного расположения и степени неправильной дифференциации серого вещества. [1]
Нарушения миграции нейронов обычно делятся на три группы:
- лиссэнцефалия / гетеротопия подкоркового пучка
- булыжник
- "Прочие" гетеротопии [3]
«Другие» типы связаны с мозолистое тело агенезия или гипоплазия мозжечка в то время как лиссэнцефалии из булыжника связаны с заболеваниями глаз и мышц. [3]
Классическая лиссэнцефалия, также известная как тип I или генерализованная агирия-пахигирия, представляет собой серьезную аномалию развития головного мозга с гладкой поверхностью мозга, аномально толстой (10-20 мм) корой с четырьмя слоями, широко распространенной гетеротопией нейронов, увеличенной желудочки и агенезия или порок развития мозолистого тела. [4] [6] Классическая лиссэнцефалия может варьироваться от агирии до регионарной пахигирии и обычно присутствует вместе с гетеротопией подкорковых полос (известной как «двойная кора» для описания кольцевых полос гетеротопических нейронов, расположенных под корой). [6] Гетеротопия подкорковых полос - это порок развития, немного отличающийся от лиссэнцефалии, который теперь классифицируется по спектру полос агирии-пахигирии, потому что он состоит из спирального узора, соответствующего широким извилинам и увеличенной толщине коры. [1] Установленная схема классификации лиссэнцефалии основана на степени тяжести (1-6 степени) и градиенте. [5]
- 1 степень: генерализованная агирия
- 2 степень: различная степень агирии
- 3 степень: пахигирия различной степени
- 4 степень: генерализованная пахигирия
- 5 степень: смешанная пахигирия и гетеротопия подкорковых лент
- 6 степень: только гетеротопия подкорковых полос
- Градиент «а»: от заднего градиента к переднему
- Градиент «b»: от переднего градиента к заднему [5]
1 и 4 степени очень редки. 2 степень наблюдается у детей с Синдром Миллера-Дикера (сочетание лиссэнцефалии с дисморфическими чертами лица, висцеральными аномалиями и полидактилией). Наиболее частая наблюдаемая лиссэнцефалия, состоящая из лобно-височной пахигирии и задней агирии, относится к степени 3. [4] Еще один порок развития, который стоит упомянуть из-за его связи с пахигирией, - это полимикрогирия. Полимикрогирия характеризуется множеством маленьких извилин, разделенных неглубокими бороздами, слегка тонкой корой, гетеротопией нейронов и увеличенным желудочком, и часто накладывается на пахигирию. [4]
лечение
Поскольку пахигирия является структурным дефектом, в настоящее время не существует лечения, кроме симптоматического, особенно при сопутствующих судорогах. Другое распространенное лечение - это гастростомия (введение зонда для питания), чтобы уменьшить возможное плохое питание и повторную аспирационную пневмонию. [4]
Микроцефальный остеодиспластический примордиальный карликовость
Микроцефальный остеодиспластический примордиальный карликовость (MOPD ) тип II представляет собой аутосомное мультисистемное заболевание, включающее тяжелую пре- и постнатальную задержку роста, микроцефалия с участием Синдром Зекеля -подобный внешний вид лица и характерные изменения скелета. Обычно у пострадавших наблюдается умственная отсталость от легкой до средней степени. Этот ребенок женского пола является первенцем от не кровных родителей на 35 неделе беременности через кесарево сечение из-за задержки внутриутробного развития. У нее была задержка психомоторного развития, и в течение первых шести месяцев жизни она неоднократно госпитализировалась из-за рецидивирующих респираторных инфекций. Ее электроэнцефалография, слуховой ответ ствола мозга оценка и хромосомный анализ были относительно нормальными. МРТ головного мозга выявила утолщение коры головного мозга с небольшими и крупными извилинами, заметно выступающими во фронтальной и задней височных областях, неполное развитие Сильвианские трещины, и расширение задние рога боковых желудочков (кольпоцефалия ). Обычно с MOPD II типа связаны только легкие пороки развития головного мозга. Результаты визуализации головного мозга этого ребенка, скорее всего, представляют диффузную пахигирию, легкую форму лиссэнцефалии. Этого ребенка результаты нервного развития были мягкими по сравнению с предыдущими отчетами о четко определенной хромосома 17 -связанный и Икс -связанная лиссэнцефалия у прикованного к постели пациента с тяжелой задержкой в развитии. [8]
Лиссэнцефалия - агирия. Шизэнцефалия. Порэнцефалия. Голопрозэнцефалия
Лиссэнцефалия - агирия. Шизэнцефалия. Порэнцефалия. Голопрозэнцефалия
Лиссэнцефалия, или агирия, — редкое заболевание, которое характеризуется отсутствием извилин большого мозга и слабым развитием сильвиевой борозды; таким образом, по внешнему виду мозг при лиссэнцефалии напоминает мозг плода на 3-4-м месяце внутриутробного развития. Данное состояние, вероятно, возникает в результате нарушения миграции нейробластов на ранней стадии внутриутробного развития и обычно ассоциируется с увеличением боковых желудочков и гетеротопиями в белом веществе. Кора при этой аномалии имеет 4-слойное строение, в отличие от обычного 6-слойного, с тонким ободком перивентрикулярного белого вещества и множественной гетеротопией серого вещества, которое выявляется при микроскопическом исследовании. В клинической картине у детей — уменьшение прироста массы тела, микроцефалия, выраженная задержка психомоторного развития и тяжелые эпилептические приступы.
Часто встречаются пороки развития глаз, включая гипоплазию зрительного нерва и микрофтальм. Лиссэнцефалия может возникать как изолированное нарушение, но примерно в 15 % случаев связана с синдромом Миллера-Дикера. У детей с этим синдромом характерные черты лица, включая выступающий лоб, выраженные двусторонние височные впадины, антеверсию (смещение кпереди) ноздрей, выступающую верхнюю губу и микрогнатию. Примерно у 90 % детей с синдромом Миллера-Дикера выявляются видимые или субмикроскопические хромосомные делеции в локусе 17р13.3 (ген LIS-1 — лиссэнцефалия 1). КТ и МРТ в типичных случаях демонстрируют гладкий мозг и отсутствие борозд.
Шизэнцефалия
Шизэнцефалией называют уни- или билатеральную расщелину в паренхиме полушарий мозга, образующуюся в результате нарушения морфогенеза. Расщелина может быть сомкнутой или разомкнутой. В случае односторонней шизэнцефалии большого размера ее можно принять за порэнцефалическую кисту. Нередко края расщелины окружены аномальным участком мозга, особенно — микрогирии. КТ служит основным методом диагностики и позволяет определить размер и протяженность расщелины. У многих пациентов отмечается тяжелая умственная отсталость в сочетании с трудно контролируемыми эпилептическими приступами и микроцефалия в сочетании со спастическим тетрапарезом при билатеральной шизэнцефалии.
![порэнцефалия]()
Порэнцефалия
Порэнцефалией называют кисту или полость в паренхиме мозга, образующуюся в результате нарушения развития или вследствие приобретенного поражения, включая инфаркт вещества мозга. Истинная порэнцефалическая киста чаще локализуется в области сильвиевой борозды и в типичных случаях сообщается с субарахноидальным пространством и/или желудочковой системой. Этот порок развития возникает в результате нарушения клеточной миграции и часто ассоциируется с другими пороками развития мозга, включая микроцефалию, аномальный паттерн прилежащих извилин и энцефалоцеле.
У детей с этим пороком развития выявляются многочисленные неврологические нарушения, в том числе умственная отсталость, спастический тетрапарез, атрофия зрительных нервов и эпилептические приступы. Псевдопорэнцефалическая киста, как правило, формируется во время пери- или постнатального периода и возникает в результате нарушений (инфаркт, кровоизлияние) артериального или венозного кровообращения. Порэнцефалические кисты, как правило, унилатеральные. Они не сообщаются с ликворсодержащими полостями и несочетаются с аномалией клеточной миграции или другими пороками развития ЦНС.
У детей с парэнцефалическими кистами на первом году жизни развиваются гемипарез и фокальные эпилептические приступы.Голопрозэнцефалия
Голопрозэнцефалия - порок развития мозга возникает в результате нарушения расщепления прозэнцефалона. Тяжесть этой аномалии может быть различной; выделяют три ее группы: алобарная, полулобарная и лобарная голопрозэецефалия в зависимости от степени тяжести аномалий расщепления прозэнцефалона. 4-й тип аномалии — срединное межполушарное сращения или синтелэнцефалия — представляет собой порок развития, при котором расщепление прозэнцефалона отсутствует в области задних отделов лобных и теменных долей. Лицевые аномалии, включая циклопию (синофтальм), цебоцефалию, агенезию резцовой (межчелюстной) кости, в тяжелых случаях встречаются часто, так как прехордальная мезодерма, дающая начало прозэнцефалону, также ответственна за индукцию срединных структур лица.
Наиболее тяжелая форма — алобарная голопрозэнцефалия, как правило, сочетается с пороками нейрональной миграции. При алобарной голопрозэнцефалии выявляются одиночный желудочек, отсутствие серпа и сращение базальных ганлиев. Этот порок развития характеризуется высокий летальностью у детей, хотя некоторые дети с этой аномалией живут в течение многих лет. При менее тяжелой голопрозэнцефалии летальность и частота тяжелых осложнений значительно ниже, варьирует в широких пределах и трудно предсказуема. Частота голопрозэнцефалии колеблется от 1/5000 до 1/16 000. В наиболее тяжелых случаях голопрозэнцефалии возможна пренатальная диагностика при УЗИ после 10 нед. гестации. Причина голопрозэнцефалии обычно неизвестна, однако во многих случаях существует связь с диабетом у матери.
В небольшой части случаев выявляются хромосомные аномалии, включая делеции хромосом 7q, Зр, 21q, 2p, 18p, 13q, а также трисомия 13 и 18. Отмечалось, что мутации гена на хромосоме 7q, кодирующего синтез протеина sonic hedgehog (SHH), служили причиной развития голопрозэнцефалии.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Аномалии головного мозга
2. Наиболее часто встречающие аномалии развития головного мозга
3. Аномалия Арнольда-Киари.
Смещение миндалин на 0,5 см и ниже у детей 5-15
лет не должно рассматриваться как патология!
Киари I – пролабирование миндалин мозжечка на 0,2-0,9
см.
Киари II – расширение большого затылочного отверстия;
низкое расположение мозжечкового намета и ствола
мозга; мост обычно сужен в переднезаднем направлении,
продолговатый мозг удлинен и опущен в позвоночный
канал; IV-ый желудочек сужен, опущен, редко изолирован
и увеличен в размерах; четверохолмная пластинка имеет
клювовидную форму; в 80-90% наблюдается дисплазия
мозолистого тела (гипоплазия или отсутствие валика,
агенезия rostrum, дисплазия falx; расширение massa
intemedia.
Киари III – крайне редкое патологическое состояние.
Грыжевое выпячивание содержимого ЗЧЯ через spina bifida
на уровне С1-С2.5. Аномалия Арнольда-Киари II, ассоциированная с полимикрогирией
6. Агенезия мозолистого тела (типичная КТ, МР-картина)
Агенезия мозолистого
тела (типичная КТ, МРкартина)
Широко расставленные
передние рога и тела боковых
желудочков.
Высокое стояние 3-го желудочка между ними.
Медиальные стенки боковых желудочков приобретают
параллельный ход.
Задние рога боковых желудочков обычно расширены.
По сагиттальным томограммам можно судить о
степени недоразвития мозолистого тела: частичная –
гипогенезия; полная - агенезия; частичный дефект
его формирования – дисгенезия.7. Частичная агенезия мозолистого тела
8. Перикаллезная липома
9. Липома четверохолмной цистерны
10. Краниостеноз, дисгенезия мозолистого тела
11. Незаращение листков прозрачной перегородки
12. Киста промежуточного паруса
13. Гипоплазия мозжечка
14. Мальформация Dandy-Walker.
Dandy-Walker, тип I. Истинная киста. Характеризуется расширением
4-го желудочка с отсутствием отверстий Можанди и Люшка,
частичной или полной агенезии червя, высоким расположением
мозжечкового намета, гидроцефалией. Связь между 4-ым
желудочком и перемедуллярным пространством отсутствует.
Dandy-Walker, тип II. Частичная агенезия червя мозжечка, связанная
с задним расширением tela chorioidea позади и над рудиментом червя.
Может сочетаться с полимикрогирией, гетеротопией, неправильным
положением ствола мозга, агенезией мозолистого тела.
Вариант Dandy-Walker . Широкая сообщаемость 4-го желудочка с
основной цистерной мозга, последняя увеличена в размерах; червь и
полушария мозжечка гипопластичны; 3-ий и боковые желудочки
гидроцефальны.15. Вариант Денди-Уолкера
16. Кистозная трансформация IV-го желудочка
17. Нарушение дивертикуляции. Голопрозэнцефалия.
18. Септо-оптическая дисплазия, дисгенезия мозолистого тела и межполушарная арахноидальная киста в левой затылочной области
19. Нарушение в формировании борозд, извилин, миграции нейронов.
Недоразвитие борозд, извилин.
Агирия – отсутствие извилин на поверхности мозга - полная
лиссэнцефалия.
Пахигирия - наличие широких и плоских извилин (неполная
лиссэнцефалия).
Шизэнцефалия (агенетическая порэнцефалия).
Врожденная расщелина, обычно идущая вдоль врожденных щелей
мозга – латеральная, центральная.
Лиссэнцефалия.
Кортикальная дисплазия (полимикрогирия).
Формирование большого количества маленьких извилин.
Излюбленная локализация – Сильвиева щель.
Гетеротопия:
Виды:
Субэпендимарная.
Фокальная (узловая).
Диффузная (лентовидная).
Смешанная.20. Лисэнцефалия
21. Агирия и агенезия мозолистого тела
22. шизэнцефалия
23. Полимикрогирия лобных долей
24. Правосторонняя мегалэнцефалия
25. Порэнцефалия левого бокового желудочка
26. фокальная кортикальная дисплазия
27. Гетеротопия (единичный узел)
28. смешанная форма гетеротопии – субэпендимарная+узловая
29. Лентовидная гетеротопия
30. платибазия
31. Наиболее типичные локализации арахноидальных кист
32. Арахноидальная киста правой височной области
33. Арахноидальная киста конвекситальной поверхности левой теменной области
34. кистозная трансформация правой теменной доли
35. Гипоплазия мозжечка
36. Аномалия Арнольда-Киари.
Аномалия АрнольдаКиари.
Смещение миндалин на 5 мм и ниже у детей 5-15 лет
не должно рассматриваться как патология!
Киари I – пролабирование миндалин мозжечка 2-9 мм.
Киари II – расширение большого затылочного
отверстия; низкое расположение мозжечкового намета
и ствола мозга; мост обычно сужен в переднезаднем
направлении, продолговатый мозг удлинен и опущен в
позвоночный канал; IV-ый желудочек сужен, опущен,
иногда изолирован и увеличен в размерах;
четверохолмная пластинка имеет клювовидную форму;
в 80-90% наблюдается дисплазия мозолистого тела
(гипоплазия или отсутствие валика, агенезия rostrum),
расширение massa intemedia.
Киари III – крайне редкое патологическое состояние.
Грыжевое выпячивание содержимого ЗЧЯ через spina
bifida на уровне С1-С2.37. Аномалия Арнольда-Киари
38. цефалоцеле
Дифференцируют по локализации дефектов костей
черепа, через которые пролабируют.
1. окципитальные.
2. свода черепа: интерфронтальные, переднего
родничка, интерпариетальные, заднего родничка,
темпоральные.
3. фронто-базилярные: фронто-назальные,
этмоидальные, назо-фронтальные, назо-этмоидальные,
назо-орбитальные.
4. базилярные: трансэтмоидальные,
сфеноэтмоидальные, транссфеноидальные, фронтосфеноидальные, сфено-орбитальные.
5. краниошизис: краниальный – верхняя фациальная
расщелина, базилярный - нижняя фациальная
расщелина, цервико-окципитальная расщелина.39. Агенезия мозолистого тела
Широко расставленные передние рога и тела
боковых желудочков.
Высокое стояние 3-го желудочка между ними.
Медиальные стенки боковых желудочков
приобретают параллельный ход.
Задние рога боковых желудочков обычно
расширены.
По сагиттальным томограммам можно судить о
степени недоразвития мозолистого тела:
частичная – гипогенезия; полная - агенезия;
частичный дефект его формирования –
дисгенезия.
Отделы мозолистого тела: rostrum, genu (колено),
body (тело), splenum (валик).40. Частичная агенезия мозолистого тела
41. Незаращение листков прозрачной перегородки
42. Мальформация Dandy-Walker.
Dandy-Walker, тип I. Истинная киста. Характеризуется
расширением 4-го желудочка с отсутствием отверстий Можанди
и Люшка, частичной или полной агенезии червя, высоким
расположением мозжечкового намета, гидроцефалией. Связь
между 4-ым желудочком и перемедуллярным пространством
отсутствует.
Dandy-Walker, тип II. Частичная агенезия червя мозжечка,
связанная с задним расширением tela chorioidea позади и над
рудиментом червя. Может сочетаться с полимикрогирией,
гетеротопией, неправильным положением ствола мозга,
агенезией мозолистого тела.
Вариант Dandy-Walker . Широкая сообщаемость 4-го
желудочка с основной цистерной мозга, последняя увеличена в
размерах; червь и полушария мозжечка гипопластичны; 3-ий и
боковые желудочки гидроцефальны.43. МР картина мальформации Dandy-Walker.
МР картина мальформации DandyWalker.
Кистозно расширенный 4-ый желудочек,
заполняющий большую часть объема ЗЧЯ.
Червь не определяется или резко
гипопластичен.
Полушария мозжечка раздвинуты и
уменьшены в объеме.
Мозжечковый намет расположен высоко.
3-и и боковые желудочки гидроцефальны.44. Нарушение в формировании борозд, извилин, миграции нейронов.
Лиссэнцефалия.
Недоразвитие борозд извилин Агирия – отсутствие извилин на
поверхности мозга полная лиссэнцефалия). Пахигирия - наличие
широких и плоских извилин (неполная лиссэнцефалия).
Шизэнцефалия (агенетическая порэнцефалия).
Врожденная расщелина, обычно идущая вдоль врожденных щелей
мозга – латеральная, центральная.
Кортикальная дисплазия (полимикрогирия).
Формирование большого количества маленьких извилин.
Излюбленная локализация – Сильвиева щель.
Виды гетеротопии:
Субэпендимарная.
Фокальная (узловая).
Диффузная (лентовидная).
Смешанная.45. гетеротопия
46. гетеротопия
47. платибазия
48. Арахноидальные кисты
49. Наиболее типичные локализации арахноидальных кист
1. киста боковой щели мозга.
2. киста конвекситальной поверхности
мозга.
3. парасагиттальная.
4. супраселлярная.
5. интраселлярная.
6. киста области вырезки мозжечкового
намета.
7. верхняя ретроцеребеллярная.
8. нижняя ретроцеребеллярная.
9. киста области мостомозжечкового угла.50. Локализация арахноидальных кист
51. Арахноидальная киста
52. Черепно-мозговая травма
53. Черепно-мозговая травма.
ПЕРВИЧНЫЕ ПОВРЕЖДЕНИЯ ГОЛОВНОГО МОЗГА
Очаговые ушибы головного мозга.
Диффузные аксональные повреждения (ДАП).
Внутричерепные кровоизлияния (эпидуральные,
субдуральные, внутримозговые гематомы).
Травматические субарахноидальные кровоизлияния.
Внутрижелудочковые кровоизлияния.
Пневмоцефалия.
Переломы свода и основания черепа.
Огнестрельные ранения головы.
ВТОРИЧНЫЕ ПОВРЕЖДЕНИЯ.
Отек мозга.
Смещения и деформации мозга (вклинения).
Сосудистые повреждения.
СМЕРТЬ МОЗГА.
ПОСЛЕДСТВИЯ ЧМТ.
Посттравматическая локальная и диффузная атрофия мозга,
энцефаломаляция, глиоз, посттравматические энцефалоцеле
и менингоэнцефалоцеле.54. Ушибы головного мозга по данным КТ и МРТ
Ушибы I типа.
Очаги гиперинтенсивного по Т2 и умерено
гипоинтенсивного по Т1 МР-сигнала,
однородной структуры.
Ушибы II типа.
Очаги гиперинтенсивного по Т2 и умеренно
гипоинтенсивного по Т1 МР-сигнала,
неоднородной структуры по Т1 за счет мелких
гиперинтенсивных включений.
Ушибы III типа.
Чередование очагов гиперинтенсивного по Т2
и умерено гипоинтенсивного по Т1 МР-сигнала,
однородной структуры и фокальных гематом.
Ушибы IV типа.
Внутримозговые гематомы.55. ДАП (диффузные аксональные повреждения).
Тяжелая ЧМТ. Характерно распределение точек
максимального повреждения вдоль трактов белого
вещества головного мозга.
Локализация 2/3 очагов субкортикальная, часто лобнопарасагиттальная, височно-перивентрикулярная, менее
часто теменная и затылочная области. Вовлечение
(всегда) мозолистого тела (валик, и задние отделы
корпуса). Могут быть внутрижелудочковые геморрагии.
Травматическое повреждение таламусов и базальных
ганглиев относительно непостоянны.
Негеморрагические.
В остром периоде – наличие небольших участков отека,
через несколько дней возможно присоединение
геморрагического компонента.
Геморрагические.
мелкие внутримозговые гематомы с типичной фазовой
трансформацией.Лиссэнцефалия - агирия. Шизэнцефалия. Порэнцефалия. Голопрозэнцефалия
Пахигирия - врожденный порок мозга, приводящий к сглаженности коры головного мозга . Как правило, дети с данным пороком имеют задержку развития и страдают от эпилепсии , начало и тяжесть проявления зависит от степени поражения коры головного мозга.
![pakhigiriya]()
Клиническая картина
Термин «пахигирия» напрямую не связан с определенной формой развития, а скорее используется для описания анатомических изменений мозга в сочетании с нарушениями миграции нейронов; чаще всего расстройства, связанные с различной степенью лиссэнцефалии. Лиссэнцефалия присутствует в 1 из 85 470 новорожденных, а продолжительность жизни у таких пациентов коротка, лишь немногие доживают до 20 лет. Пачигирия - это состояние, идентифицируемое по типу кортикальной генетической мальформации.
Связь с эпилепсией, лиссэнцефалией и подкорковой гетеротопией
Различные степени выраженности и проявления эпилепсии связаны с пороками развития коры. Исследователи полагают, что около 40% детей с диагнозом лекарственно-резистентной эпилепсии имеют некоторую степень аномалии развития коры.
Лиссенцефалия (с которой наиболее тесно связана пахигирия) связана с тяжелой умственной отсталостью , эпилепсией и двигательной недееспособностью . Две характеристики лиссенцефалии включают отсутствие извилин ( агирия ) и сглаженность извилин (пахигирия). Другие возможные симптомы лиссэнцефалии включают телекантус , эстропию , гипертелоризм , различные уровни умственной отсталости, гипоплазию мозжечка , аплазию мозолистого тела. Более 90% детей, страдающих лизенцефалией, имеют судороги.
Пациенты с субкортикальной гетеротопией (другое расстройство, связанное с пахигирией) обычно имеют более мягкие симптомы, и их когнитивная функция тесно связана с выраженностью субкортикальной гетеротопии и степенью присутствия пахигирии.
![lissencephalia-1]()
Причины
Пахигирия вызвана расстройством процесса миграции нейронов плода из-за генетических или, возможно, экологических воздействий. У пораженной коры головного мозга обычно будет только четыре развитых слоя вместо обычных шести. Одним из наиболее известных и наиболее распространенных типов нарушений миграции нейронов является лиссэнцефалия - диффузная кортикальная мальформация, относящаяся непосредственно к агирии и пахигирии.
Неполная миграция нейронов во время раннего развития мозга плода является предшественником лиссанцефалии. Если нейроны имели аномальную миграцию во время развития - возможны корковые дисплазии, включающие:
- классическая лиссанцефалия (как указано выше) и
- гетеропию в виде подкорковых полос (агирийно-пахигириевый спектр).
![lissencephalia-3]()
Патогенез
Пахигирия, лиссенцефалия (гладкий мозг) и полимикрогирия (множественные мелкие гири) - все это результаты аномальной миграции неврных клеток коры головного мозга. Аномальная миграция, как правило, связана с неорганизованной клеточной архитектоникой, отсутствием сформированных шести слоев кортикальных нейронов (обычно имеется четырехслойная кора) и функциональные нарушения. Аномальное образование головного мозга может повлечь наличие судорожных симптомов, задержку развития и умственные дисфункции .
Диагностика
Различные методы визуализации обычно используются для диагностики. Хотя компьютерная томография (КТ) обеспечивает более высокое пространственное разрешение изображения головного мозга, пороки развития коры головного мозга лучше визуализируются с использованием магнитно-резонансной томографии (МРТ), которая обеспечивает более высокую тканевую контрастность и лучшее разграничение белого и серого вещества.
Лечение
Поскольку пахигирия является структурным дефектом, в настоящее время этиопатогенетического лечения нет, кроме симптоматического лечения, особенно для пациентов с приступами эпилепсии. Другим распространенным методом лечения является гастростомия (введение питательной трубки) для снижения возможного плохого питания и повторной аспирационной пневмонии.
Полная или частичная перепечатка данной статьи, разрешается при установке активной гиперссылки на первоисточник
Если у вас остаются сомнения в выводах по результатам вашего МРТ - вы можете заказать пересмотр вашего исследования с подробной расшифровкой здесь:
Возможно вас так же заинтересует
Нейроэпителиальные кисты также называемые нейроглиальными или глио-эпендимными кистами, являются аномалиями развития, возникающими в результате поглощения части развивающейся нейроэктодермы, из лептоменингиальной нейроглиальной гетеротопии.
Расширенные пространства Вирхова-Робина появляются во всех возрастных группах. С возрастом пространства ВР обнаруживаются с большей частотой и большими кажущимися размерами.
Аномалия Киари - врожденное смещение структур задней черепной ямки в каудальном направлении.Основную роль сыграл Chiari, который в 1891 г. описал аномалии заднего мозга и дал их классификацию.
Цефалоцеле характеризуются дефектом свода или основания черепа, через которые выпячиваются структуры мозга. В зависимости от грыжевого черепного отверстия выделают менингоцеле, менингоэнцефалоцеле, атретическое целе и глиоцеле.
Агенезия мозолистого тела (АМТ) - редкое расстройство, которое присутствует при рождении (врожденное). Он характеризуется частичным или полным отсутствием (агенезией) мозолистого тела. Мозолистое тело состоит из поперечных волокон.
Внутричерепная липома представляет собой доброкачественное образование, которое состоит из жировой ткани. Данная патология преимущественно развивается бессимптомно. Липомы (жировики), не имеют склонности к раковой трансформации. Внутричерепная липома всегда врожденное образование и не связано с неоплазией, а является нарушением эмбриогенеза с формированием патологической области отложения жировой ткани.
Комплекс Денди-Уокера это полная или частичная агенезия червя мозжечка, вращение мозжечка против часовой стрелки с расширением IV желудочка на фоне расширения задней черепной ямки за счет смещения намета вверх.
Ромбэнцефалосинапсис - аномалия развития ромбовидного мозга (rhombencephalon). В состав ромбовидного мозга входит: мозжечок, мост и продолговатый мозг, окружающие ромбовидную ямку, которая является дном 4-го желудочка.
Голопрозэнцефалия - порок развития с нарушением формирования переднего мозга (prosencephalon), на ранних стадиях развития плода, на 3й неделе гестации (когда существуют 3-5 мозговых пузырей) при котором передний мозговой пузырь не разделен частично или полностью на два симметричных полушария.
Септооптическая дисплазия (синдром de Morsier) является одной из форм лобарной голопрозэнцефалий. Характеризуется отсутствием прозрачной перегородки, гипоплазией зрительных нервов, хиазмы и воронки.
Шизэнцефалия - расщепление коры головного мозга линейной формы или широким (но не обширным) проходом, которое распространяется от желудочков к субарахноидальному пространству.
Пахигирия - врожденный порок мозга, приводящий к сглаженности коры головного мозга . Как правило, дети с данным пороком имеют задержку развития и страдают от эпилепсии , начало и тяжесть проявления зависит от степени поражения коры головного мозга.
Изменение величины мозга - врожденная асимметрия размеров мозговых структур. Микроцефалия, или микроэнцефалия - аномально маленький головной мозг. Обычно это является результатом внутриутробного сосудистого инсульта или внутриутробной TORCH-инфекции, также причиной могут быть метаболические нарушения, в частности фенилкетонурия.
Порэнцефалия (porencephalia, греческий: poros - проход, отверстие, пора + enliephalos - головной мозг) — патологическое кистозное расширение полости желудочка головного мозга разной величины, сообщающееся с субарахноидальным пространством. В действительности граница между порэнцефалией и шизэнцефалией смазана.
Нейрофиброматоз I типа (болезнь Реклингхаузена) — наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Впервые описано во второй половине XIX века в 1882 году учеником Рудольфа Вирхова - Фридрихом фон Реклингхаузеном.
Нейрофиброматоз II типа — наследственное заболевание с аутосомно-доминантным типом передачи, которое наследуется или возникает спонтанно, характеризующееся образованием множественных доброкачественных опухолей, преимущественно шванном и менингиом, локализующихся в центральной нервной системе и по ходу периферических нервов.
Энцефало-тригеминальный ангиоматоз (болезнь Стерджа-Вебера, синдром Штурге-Вебера или синдром Стерджа-Вебера-Краббе) — спорадически возникающее заболевание, с возникновением ангиом мягкой мозговой оболочки и кожи лица, как правило в области глазной и верхнечелюстной ветвей тройничного нерва.
Туберозный склероз (болезнь Бурневилля-Прингла) - генетически детерминированное заболевание, характеризующееся выраженным ростом глии в ткани мозга и сетчатки, а так же гиперплазией производных экто- и мезодермы, поражением кожи, нервной системы и наличием доброкачественных опухолей (гамартом) в различных органах.
Болезнь Гиппеля—Линдау (цереброретинальный ангиоматоз) — факоматоз, при котором гемангиобластомы мозжечка сочетаются с ангиомами спинного мозга, множественными врождёнными кистами поджелудочной железы и почек. У четверти больных развивается карцинома почки, часто первично-множественная.
Гамартома гипоталамуса - это редкое неопухолевое образование гипоталамуса. По морфологической классификации соответствует ганглиоцитоме (доброкачественная опухоль из элементов симпатических нервных ганглиев). На сегодня считается, что данное образование не представляет сложности в диагностики, но затрудняет лечение.
Общая схема форм черепа . Мезоцефалический череп (Мезокран) - нормальная анатомическая форма черепа, средняя статистически встречаемая форма - стандартная форма черепа. Долихоцефалический череп (Долихоцефал) - значительное преобладание передне-заднего размера черепа над фронтальным размером.
Киста шишковидной железы - доброкачественный вариант нормальной шишковидной железы. Киста шишковидной железы отличается от опухоли равномерной тонкой стенкой (до 0,2см) в которой могут быть петрификаты. Содержимое кисты может быть изоинтенсивна ликвору или серому веществу, а так же может быть гиперинтенсивна по Flair (что связано с отсутствием движения ликвора в кисте, а так же увеличенным количеством белка или дериватами гемоглобина).
Лиссэнцефалия - агирия. Шизэнцефалия. Порэнцефалия. Голопрозэнцефалия
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Ваша заявка принята!
Благодарим за обращение.
В ближайшее время с вами свяжется наш специалист.
Лиссэнцефалия, шизэнцефалия, порэнцефалия: причины, симптомы, прогноз
Сглаживание извилин коры головного мозга (агирия) формируется внутриутробно. Патогенетическим механизмом развития нозологии является нарушение эмбриогенеза, обусловленного патологией распространения из первичной нервной трубки нейробластов. Сглаживание поверхности сопровождается снижением умственной, интеллектуальной активности.
Сопутствующие неврологические расстройства постепенно нарастают. Заболевание приводит к летальному исходу еще в грудном возрасте.
Отсутствие мозговых извилин – исключает оптимальное функционирование второй сигнальной системы, которая существует только у человека. Складчатость повышает функциональную поверхность мозга.
Даже раннее выявление с помощью МРТ головного мозга не позволяет спасти жизнь ребенку с лиссэнцефалией первого или второго типа.
На протяжении беременности врожденные пороки развития церебральных структур определяются ультразвуковым обследованием (УЗИ). Достоверность процедуры – 60%, поэтому иногда после МРТ выявляются врожденные пороки развития мозга, хотя внутриутробный мониторинг патологии не определяет патологических состояний.
Пример заключения магнитно-резонансной томографии:
- Билатеральная вентрикуломегалия;
- Увеличение ликворных пространств;
- Сглаженность церебральных извилин;
- Неполное отсутствие складок теменных и лобно-височных областей;
- Расширение боковых желудочков.
Нозология нередко сочетается со стигмами церебрального дисэмбриогенеза – макроцефалией, агирией, микроцефалией.
Медицинские источники утверждают, что патологию может обнаружить УЗИ после двадцать шестой недели беременности.
![lissencefaliya]()
МРТ: норма и лиссэнцефалия
Лиссенцефалия 2 типа имеет альтернативное название «синдром Норман-Робертс». Канадские ученые, открывшие заболевание, считают основной причиной формирования заболевания мутацию гена «RELN».
Участок ДНК, отвечающий за нозологию, кодирует образование протеина «рилина». Синдром характеризуется множеством клинических изменений:
- Лимфедема (лимфатический отек);
- Эпилептические приступы;
- Покатость лба;
- Утолщение церебральной коры;
- Агирия (сглаженность мозговых борозд);
- Макроцефалия;
- Микроцефалия.
Перинатальное УЗИ обнаруживает большую часть случаев болезни.
Причины возникновения лиссенцефалии
Формирование внутримозговой коры у плода проходит несколько стадий развития. Вначале происходит деление нейронов. Постепенно нервные клетки мигрируют из первоначального матрикса в места постоянного расположения. Остальные этапы эмбриогенеза сопровождаются специализацией клеточных пластов, отвечающих за анатомические структуры.
Сложный процесс образования мозга должен проходить под строгим контролем биохимических механизмов. Любое внешнее вмешательство нарушает внутриутробное развитие.
Основные причины возникновения лиссэнцефалии:
- Внутриутробная гипоксия (кислородное голодание);
- Вирусные инфекции;
- Хромосомные аномалии (дефекты формирования протеина рилина в 7-ой хромосоме).
Внутрисемейные случаи передачи заболевания достаточно редки. Если встречается нозология у нескольких членов семьи, требуется генетическое консультирование.
Средняя распространенность нозологии первого типа – 11% на миллион новорожденных детей. Большинство ситуаций провоцируется генетическими дефектами гена LIS1. На втором месте встречается гипоплазия мозжечка из-за дефекта гена TUBA1A.
После наследственных аномалий на втором месте – вирусное инфицирование в первом триместре беременности.
Основные виды лиссенцефалии
Встречается свыше двадцати морфологических форм заболевания:
- Классическая форма (I типа) с мутацией LIS1, синдромом Миллера-Дикера;
- Форма с наследственным дефектом DCX;
- Изолированная разновидность без мутаций;
- Агенезия мозолистого тела при аномалии Х-хромосомы;
- Гипоплазия мозжечка с нарушением кодирования рилина (синдром Нормана-Робертса);
- Микролиссэнцефалия;
- Булыжниковый вид с синдромом Фукуямы, Уокера-Варбурга, мышечно-глазо-мозговой формой.
Мышечная дистрофия с олигофренией Фукуяма передается аутосомно-рецессивным способом. Наследственная миотония имеет прогрессирующее течение.
Особенности шизэнцефалии, порэнцефалии, голопрозэнцефалии
Билатеральная расщелина церебральных полушарий формируется на этапе эмбриогенеза. Морфологическая структура образования бывает разомкнутой или сомкнутой.
Односторонняя локализация патологии на МРТ снимках может напоминать кисту. Вдоль расщелины расположены патологические участки церебральной паренхимы – микрогирия, зоны повышенной повторной активности, спастические параличи.
Порэнцефалия характеризуется образованием кистозных полостей внутри церебральной паренхимы. Причины возникновения патологии – кровоизлияние внутрь головного мозга, инфаркты, инсульты. Типичная локализация кисты – сильвиева борозда.
В редких случаях порэнцефалия характеризуется образованием аномальных ходов между кистозной полостью и желудочковыми пространствами. Нозология нередко характеризуется другими стигмами дисэмбриогенеза:
- Микроцефалия;
- Энцефалоцеле;
- Агирия.
У ребенка с пороком развития возникают неврологические расстройства:
- Олигофрения (умственная отсталость);
- Приступы эпилепсии;
- Атрофические изменения дисков зрительного нерва;
- Тетрапарез.
Осложняют клиническую картину сопутствующие изменения:
- Артериальные и венозные кровоизлияния;
- Внутричерепная гипертензия (при нарушении циркуляции ликвора);
- Церебральные инфаркты;
- Гемипарезы;
- Очаговые эпилептические очаги.
Псевдопорэнцефалические кисты нередко имеют унилатеральное (одностороннее) расположение. Нередко сочетаются с пороками центральной нервной системы, дефектами клеточной миграции.
![shizzencefaliya]()
Шизэнцефалия на МРТ
Голопрозэнцефалия – что это такое
Заболевание возникает по причине патологического разделения химического вещества прозэнцефалона.
Классификация нозологии по степени тяжести:
- Лобарная;
- Полулобарная;
- Алобарная.
Самая тяжелая – последняя форма. При ней наблюдается ряд врожденных аномалий:
- Недоразвитие межчелюстной кости;
- Синофтальм;
- Цебоцефалия.
На фоне множественных аномалий части лица трудно различимы. Сопутствующие пороки нейрональной миграции:
- Синостоз (сращение нервных ганглиев);
- Наличие одного церебрального желудочка.
Этиологические механизмы наследственных дефектов не установлены. Голопрозэнцефалия в большинстве случаев завершается смертельным исходом, но неполная форма может приводить к инвалидности.
Симптомы лиссэнцефалии
Клинические проявления нозологии определяются сразу после рождения ребенка. Отмечаются не только генетические дефекты, но и нарушения глотания у малыша, повышенный мышечный тонус, отсталость в развитии от сверстников.
Симптомы лиссэнцефалии у ребенка 2-5 месяцев:
- Олигофрения;
- Нарушение умственного развития;
- Миоклония;
- Увеличение артериального давления.
Вначале умственная отсталость не прослеживается. После первого года жизни наблюдаются расстройства психоречевых навыков. Дети раздражительны, тревожны, мышечная активность снижена.
При сравнении со сверстниками наблюдается значительное отставание в развитии, формировании мышечной системы, иннервации внутренних органов, малого таза.
При легком течении заболевания ребенок может дожить до подросткового возраста. Внешние аномалии ограничивают социального общения. Больные редко доживают до восемнадцати лет, но существует около двадцати разновидностей патологии. Проявления первого типа:
- Макрогирия;
- Шизэнцефалия;
- Микроцефалия;
- Четырехслойное строение коры;
- Гипоплазия моста;
- Недоразвитие мозжечка.
Определить лиссэнцефалию при отсутствии положительных результатов внутриутробного УЗИ сложно. Клиническая картина возникает после рождения. Мышечные параличи, судороги появляются сразу или на протяжении первого года.
В большинстве случаев установить диагноз лиссэнцефалии поможет ультразвуковое обследование. Врач может назначить магнитно-резонансную томографию беременной женщине и ребенку. МРТ является безвредной, но в первом триместре не проводится из-за отсутствия практической информации о влиянии магнитного поля на плод.
Первые признаки лиссэнцефалии у новорожденных
Раннее выявление нозологии помогает продлить жизнь малышу, но спасти от смерти не удается. Первые признаки у новорожденных:
- Небольшая голова;
- Апатия, вялость;
- Широкое межглазное расстояние;
- Эпилептический судорожный синдром;
- Гипертонус мускулатуры;
- Увеличение ширины между легкими и почками;
- Активация патологических рефлексов;
- Покатая лобная часть.
На протяжении первого года жизни присоединяются другие проявления – речевые расстройства, недержание мочи, учащение дыхания и сердцебиения.
Синдром летального птеригума (Нормана-Робертса) возникает при лиссэнцефалии первого типа. К вышеописанным проявлениям присоединяется ряд проявлений:
- Сложности сидения;
- Невозможность удержания малышом вертикальной позиции;
- Появление генерализованных подергиваний мускулатуры;
- Аномалии краниофациальной области (широкие глаза, покатый лоб, бугры на затылке);
- Глазной нистагм;
- Снижение мышечного тонуса.
Мозжечковая атаксия характеризуется нарушением координации. Ребенок не может удержать равновесия, поддерживать вертикальную позицию. Родители вначале обращают внимание на сложности посадки, сохранения горизонтального положения.
Диагностика лиссэнцефалии
Устанавливается диагноз сразу после рождения с помощью УЗИ, КТ и МРТ. Внутриутробно обнаружить проявления можно с 20 недели. Выявление малейших нарушений развития головного мозга плода требует дополнительной диагностики. В зависимости от степени повреждений принимается решение о возможности прерывания беременности. При подозрении на наследственные формы проводится генетическое консультирование всех членов семьи.
Поздняя верификация патологии требуется постоянного консервативного лечения. Терапия тяжелых форм проводится в стационаре под контролем квалифицированных врачей.
Основные виды диагностики лиссэнцефалии:
- Магнитно-резонансная томография (МРТ головы) показывает мягкие ткани. Исследование обнаруживает церебральные аномалии, кисты, скопления жидкости, воспалительные очаги;
- Внутриутробное УЗИ головы выполняется на 22-27 неделе, когда прослеживается процесс образования борозд;
- Компьютерная томография (КТ) помогает верифицировать изменения серого и белого вещества, дополнительные твердые образования. Обследование приводит к радиационному облучению тканей, поэтому детям выполняется по строгим показаниям;
- Электроэнцефалография (ЭЭГ) верифицирует очаги повышенной мозговой активности, участки гиперактивности паренхимы.
Самое достоверное исследование – МРТ при лиссэнцефалии у новорожденных, позволяющее определить самые мельчайшие аномалии.
Дифференциальная диагностика
Схожими клиническими признаками характеризуется ряд хромосомных аномалий – синдром Пена-Шокейра, Нунана, артрогрипоз. Генетическое консультирование верифицирует нозологии до появления клинических симптомов.
Контрактуры суставов удается диагностировать ближе к первому году, когда ребенок предпринимает попытки сидеть или ходить. Внешние проявления водянки мозги – увеличенная форма черепа, выпячивание родничков.
Прогноз и продолжительность жизни
Большинство детей не доживает до года. Если ребенку удается дожить до 3 лет, он остается инвалидом и погибает от сопутствующих инфекций, тяжелых осложнений – парезы кишечника, мочевого пузыря, конечностей, дыхательной мускулатуры.
Угнетение глотательного рефлекса обуславливает затруднения питания, что обеспечивает дефицит массы тела, приводит к снижению иммунитета.
Прогноз неблагоприятный даже при внутриутробном выявлении аномалий развития.
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Читайте также: