Субкортикальная гетеротопия: лиссэнцефалия
Обновлено: 18.04.2024
Панель "Нейродегенеративные заболевания"
Нейродегенеративные заболевания представляют собой широкий спектр различных по своей природе болезней, обусловленных постепенной гибелью отдельных групп нервных клеток и характеризующихся неуклонно прогрессирующим неврологическим дефицитом, включая двигательные расстройства, психоэмоциональные и когнитивные (вплоть до деменции) нарушения и эпилептические приступы. Необходимо отметить, что нейродегенеративные заболевания могут манифестировать как во взрослом, так и в детском возрасте.
Эпидемиология
Более 40 млн людей по всему миру страдают различными нейродегенеративными заболеваниями, из них порядка 10 млн — это пациенты с болезнью Паркинсона. В раннем возрасте распространённость нейродегенеративных заболеваний составляет около 1:1500.
Механизмы развития нейродегенеративных заболеваний
Патофизиологические механизмы нейродегенеративных заболеваний включают в себя синтез и агрегацию белков с патологической конформацией, что, в свою очередь, запускает каскад иммунных и метаболических нарушений, накопление металлов в центральной нервной системе, нарушение энергетического обмена клеток, дефицит ферментов и т.д. При этом, большая роль в развитии нейродегенеративного процесса принадлежит генетическим факторам. Выделяют как моногенные нейродегенеративные заболевания (например, болезнь Гентингтона, спинальные амиотрофии, ряд вариантов спиноцеребеллярных атаксий, болезнь Вильсона, моногенные варианты болезни Паркинсона, спастические параплегии, пароксизмальные дискинезии и пр.), так и болезни, развивающиеся вследствие нарушений функционирования целых ансамблей генов в сочетании с действием неблагоприятных факторов внешней среды (идиопатическая болезнь Паркинсона, атипичный паркинсонизм, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височная деменция и пр.). При этом, важно помнить, что отсутствие отягощённого семейного анамнеза по нейродегенеративному заболеванию не исключает наличие генетической предрасположенности к его развитию.
Диагностика
Диагностика нейродегенеративных заболеваний складывается из комплексной оценки клинической картины, характера дебюта и прогрессирования заболевания, лабораторно-инструментальных исследований и данных нейровизуализации. Отдельное место занимают в диагностическом процессе методы генетического тестирования. Учитывая сложные взаимоотношения генотипа и фенотипа заболевания, очень часто генетическое тестирование не ограничивается анализом одного-двух генов. В трудных диагностических случаях для удешевления и повышения эффективности диагностического поиска широко применяется метод высокопроизводительного секвенирования, позволяющего анализировать сразу до нескольких сотен генов.
- Деменции (болезнь Альцгеймера, лобно-височная деменция, наследственные прионные заболевания).
- Паркинсонизм (болезнь Паркинсона, мультисистемная атрофия, кортикобазальная дегенерация, болезнь диффузных телец Леви, прогрессирующий надъядерный паралич).
- Наследственные мышечные дистонии.
- Пароксизмальные двигательные расстройства (пароксизмальные дискинезии).
- Нейродегенерация, ассоциированная с накоплением металлов (болезнь Вильсона, нейродегенерации с накоплением железа, синдром Фара, накопление марганца).
- Атаксии (спиноцеребеллярные атаксии, не связанные с экспансией нуклеотидных повторов).
- Наследственные моторно-сенсорные нейропатии (болезнь Шарко-Мари-Тута).
- Нейрометаболические заболеваний (нейрональный цероидный липофусциноз, лизосомные болезни накопления, сфинголипидозы).
- Спастические параплегии (болезнь Штрюмпеля, в т.ч. осложнённые формы).
- Болезни мотонейрона (боковой амиотрофический склероз, спинальные мышечные атрофии).
- Болезни белого вещества (синдром исчезающего белого вещества, прогрессирующие лейкодистрофии).
- Эпилептические энцефалопатии.
- Редкие моногенные синдромы, сопровождающиеся нейродегенерацией (болезнь Айкарди-Гутьерес, синдром Жубер, синдром Кноблоха).
- Митохондриальные заболевания вследствие мутаций в ядерных генах.
Что даёт генетическое исследование?
Завершение диагностического поиска.
Выявление риска передачи заболевания по наследству. Благодаря генетической диагностике можно просчитать риск заболевания в последующих поколениях и избежать повторных случаев в семье.
Подбор правильного лечения. Несмотря на то, что в настоящее время нейродегенеративные заболевания невозможно излечить, ряд из них (например, болезнь Паркинсона, болезнь Вильсона, ряд заболеваний вследствие нарушений метаболизма) имеют специфическое патогенетическое и симптоматическое лечение. Например, при болезни Вильсона применяется пеницилламин, при болезни Ниманна-Пика типа С — миглустат. Правильный диагноз является залогом верно назначенного и эффективного лечения.
Описание и возможности метода
Исследование проводится с помощью метода высокопроизводительного секвенирования (NGS) на секвенаторе нового поколения со средним покрытием не менее 70-100х. В панель «Нейродегенеративные заболевания» входят 723 гена, мутации в которых могут приводить к развитию нейродегенеративных заболеваний. Не стоит забывать, что некоторые заболевания из группы наследственных НДЗ обусловлены экспансией нуклеотидных повторов или хромосомными перестройками, что является ограничением данного метода.
При проведении процедуры строго соблюдается конфиденциальность.
Пациент получает точные результаты, на основании которых специалисты могут составить впечатление о молекулярно-генетическом диагнозе и максимально эффективно скорректировать лечение, а также провести медико-генетическое консультирование.
Как пройти исследование
Подготовка к анализу: нет
Дни забора (приема) материала: по графику работы медицинского центра Примечание: Необходимо иметь с собой направление и выписки/заключения от врачей
Нарушения развития коры головного мозга
При МРТ в СПб мы прицельно ищем аномалии коры головного мозга при эпилепсии и отставании в развитии. Нарушения развития коры может быть изолированной аномалией развития, либо сочетаться с другими нарушениями развития, такими как нарушения регионализации. Нарушения развития коры определяются при МРТ головного мозга и могут быть разделены на:
Нарушения пролиферации и дифференциации – микроцефалия, мегалэнцефалия
Нарушения миграции – агирия-пахигирия (лиссэнцефалия), полимикрогирия, гетеротопии
Нарушения организации коры – микродисгенезии
Мегалэнцефалия представляет собой увеличение одного или обоих полушарий мозга. При мегалэнцефалии при МРТ наблюдается увеличенный боковой желудочек с соответствующей стороны, кора утолщена и не разделена на извилины (агирия), белое вещество не миелинизировано.

МРТ. Т1-зависимая корональная томограмма. Агирия.
Гетеротопии. В ходе эмбриогенеза нейроны могут не достичь своего места в коре. Большинство нарушений миграции имеет доминантное, связанное с Х-хромосомой, происхождение. Аномалии могут быть локальными и диффузными. Диффузные гетеротопии локализуются перивентрикулярно. Серое вещество по данным МРТ скапливается только вокруг боковых желудочков, не затрагивая области вокруг III и IV желудочков. В четверти случаев гетеротопии сопутствуют аномалии мозолистого тела и мозжечка.
Если нейроны совсем не достигают коры, то возникает лиссэнцефалия. Если только часть нейронов не достигает её, то возникают субкортикальные гетеротопии, видимые при МРТ в виде узлов или полосы («двойная» кора). Клиническая симптоматика обычно негрубая – небольшое отставание развития, пирамидные знаки и, иногда, дизартрия.

МРТ. Т1-зависимая аксиальная томограмма. “Двойная кора”.
Очаговые (фокальные, узловые) гетеротопии принято ещё называть гамартомами. Они встречаются как самостоятельная аномалия или как проявление туберозного склероза. На МРТ сигнал от узлов типичный для серого вещества и они типично не контрастируются гадолинием. Это позволяет отличать их от субэпендимальных узлов при туберозном склерозе. Особым вариантом гамартомы является гипоталамическая гамартома. Она расположена в области серого бугра, между ножкой гипофиза и сосочковыми телами. Гипоталамическая гамартома имеет экзофитный тип роста и достигает 12 мм. Клинически бывает бессимптомной, либо проявляется ранним созреванием, акромегалией и особым видом парциальной эпилепсии – судорогами в виде навязчивого смеха, а также психическими нарушениями. На Т1-зависимых МРТ гипоталамическая гамартома изоинтенсивна белому веществу, на Т2-зависимых МРТ немного гиперинтенсивнее его. Образование однородное, имеет четкий контур. Масс-эффект выражается в смещении воронки гипофиза. В отличие от астроцитомы той же локализации гамартома не вовлекает перекрест зрительных нервов. Труднее отличить гамартому от менингиомы, но последняя усиливается при контрастировании. Редко встречается ганглиоглиома гипоталамуса. Она содержит кисты, иногда микрокальцинаты (что видно при КТ) и примерно в половине случаев усиливается при контрастировании. Также редко встречаются липомы гипоталамуса, которые имеют характерный для жировой ткани сигнал.
Лиссэнцефалия – это общий термин, под которым понимают нарушение формирования борозд. Крайнее проявление ее – полное отсутствие извилин – агирия. Серое вещество имеется, но оно не разделено бороздами. Агирия может быть локальной, обычно этот тип наблюдается в височной доле.
Аномально малое число извилин в связи с неполными бороздами называется пахигирией. Обычно, она также локальная, извилины широкие и сглаженные. Сочетание участков пахигирии и агирии называют лиссэнцефалией I типа. При МРТ определяется утолщение коры, вертикальные Сильвиевы борозды и часто выпрямленные гиппокампы. Клинические проявления укладываются в различные формы (синдромы Миллера – Декера, Нормана – Робертса и т.д.), проявляющиеся в первый год жизни. Тип II отличается нарушением структуры самой коры, которая пронизана сосудами и фиброглиальными пучками. Этот тип сочетается с гидроцефалией и неполной миелинизацией. Характерно клиническое проявление в виде синдрома Уокера – Варбурга.
Полимикрогирия – множественные неглубокие извилины. Часто сочетается с гетеротопией серого вещества и гемимегалэнцефалией. Считается, что патогенез полимикрогирии связан с ишемическим некрозом пятого слоя коры до 20 недели эмбриогенеза. Часть случаев связана с врождённой цитомегаловирусной инфекцией.
Кроме того полимикрогирия может входить в состав синдром Экарди (Aicardi) -Х-связанной доминантной патологией. Он протекает в виде спазмов и хориоретинопатии. При МРТ часто выявляются гипоплазия мозжечка, агенезия или недоразвитие мозолистого тела, кисты ЗЧЯ и средней линии, папилломы сосудистого сплетения.

МРТ. Т1-зависмая сагиттальная томограмма. Синдром Экарди.
МРТ. Т1-зависимая корональная томограмма. Полимикрогирия.
Микродисгенезии коры ответственны за некоторые варианты экстратемпоральной эпилепсии и, по-видимому, за некоторые психические патологии. Только иногда микродисгенезии дают изменения на макроскопическом уровне, которые выявляются при МРТ.
Корковые аномалии лучше видны в высоких полях, чем в открытом МРТ. МРТ СПб дает возможность исследовать методом МРТ в разных центрах, но столь сложные патологии мы советуем искать только в специализированных центрах.
Субкортикальная гетеротопия: лиссэнцефалия
Субкортикальная гетеротопия: лиссэнцефалия
Узловые гетеротопии серого вещества присутствуют у многих пациентов с другими нарушениями миграции, такими как полимикрогирия или шизэнцефалия. Одиночные или многоочаговые крупные гетеротопические узлы могут являться фокусами парциальных судорог. Однако даже гигантские гетеротопии, затрагивающие одно полушарие, могут оставаться бессимптомными. Детализированное нейропсихологическое исследование одного из таких случаев продемонстрировало едва уловимые нарушения полушарных функций, несмотря на нормально развитый интеллект (Calabrese et al., 1994).
Спектр классической лиссэнцефалии и субкортикальной линейной гетеротопии. Эти нарушения миграции могут быть рассмотрены для оценки различных степеней тяжести основной патологии нейрональной миграции, хотя генетически они различаются (Palmini et al., 1993).
Под лиссэнцефалией понимают гладкий мозг. Термин агирия-пахигирия лучше, так как поверхность мозга не всегда гладкая (Aicardi, 1991). В наиболее тяжелых случаях извилины не формируются (агирия). В большинстве случаев присутствует несколько извилин (пахигирия). Dobyns и Leventer (2003) различают 6 степеней лиссэнцефалии (от 1 до 6), в зависимости от количества извилин, видимых на МРТ. Только степень I заслуживает названия лиссэнцефалии; степени 2-4 являются случаями с пахигирией, и степени 5 и 6 относятся к субкортикальной линейной гетеротопии. В данном разделе объединены различные типы, как имеющие сходный спектр и, очевидно, отчасти сходные механизмы. Хотя имеется несколько форм лиссэнцефалии, в этом разделе рассмотрен только вариант мутации гена LIS1 на 17 хромосоме.

• Классическая (тип 1, Бильшовского) лиссэнцефалия. При классической лиссэнцефалии мозг имеет малые размеры и только первичные, и иногда несколько вторичных извилин. При отсутствии извилин извилистыми становятся сосуды. Кора патологически утолщена (10-20 мм), тогда как белое вещество выглядит узкой полосой вдоль желудочков. Типично наличие четырех слоев коры:
1) поверхностный, разреженный клеточный слой, аналогичный молекулярному слою нормального мозга;
2) узкий, густоклеточный слой, где располагаются большие пирамидальные нейроны, которые в норме должны располагаться в более глубоких отделах;
3) тонкий слой белого вещества, ниже которого находится
4) широкая полоса малых эктопированных нейронов, распрострающаяся почти до стенки желудочков (Dobyns и Leventer, 2003).
Многие нейроны в клеточных слоях ориентированы неправильно, с апикальным дендритом, направленным вниз или вбок (Takashima et al., 1987). Более глубокий клеточный слой сформирован из эктопированных нейронов, остановившихся на пути их миграции из герминативного слоя к коре примерно на 12 неделе гестации, поэтому кора выглядит как у 13-недельного плода. Нейроны этого слоя имеют избыточную колонковую организацию. В продолговатом мозге характерна эктопия ядра оливы. Зубчатые ядра ненормально запутанны, и пирамиды гипоплазированы или отсутствуют (Friede, 1989).
Некоторые из таких случаев являются частью специфического дисморфического близкого генного синдрома, синдрома Миллера-Дикера, который характеризуется узким лбом, широкой переносицей, отсутствием выемки верхней губы, вздернутыми ноздрями, ретрогнатизмом, аномалиями пальцев и гиперваскуляризацией сетчатки (Dobyns и Leventer, 2003). В таких случаях Dobyns и Truwit (1995) выявили явную делецию 17р13.3 у 14 из 25 пациентов и субмикроскопические делеции в 25 из 38 случаев с использованием цитогенетических методов и в 35 из 38 случаев с флюоресцентной гибридизацией in situ. Сиблинги с синдромом Миллера-Дикера рождались у пар, в которых у одного из родителей произошла пропорциональная транслокация концевого фрагмента хромосомы 17р на хромосому 13-15 пары, что проявлялось в несбалансированных формах у пострадавших детей (Greenberg et al., 1986, Dobyns и Leventer, 2003).

(слева) Тип I (классическая) лиссэнцефалия. Четырехслойная кора. От поверхности (сверху) вниз:
(1) молекулярный слой;
(2) поверхностный клеточный слой, содержащий несколько типов клеток, включая большие пирамиды, в норме располагающиеся глубже в пятом слое;
(3) широкий, бесклеточный слой;
(4) широкая полоса гетеротопированных клеток, остановленных при миграции — обратите внимание на столбчатое расположение.
(справа) Нормальное расположение.
Большинство случаев с типом I лиссэнцефалии не являются частью синдрома Миллера-Дикера и определяются как «изолированное» последствие лиссэнцефалии.
Клинические проявления во всех случаях отличаются тяжелой задержкой умственного развития и диплегией, часто атонического типа (de Rijk-van Andel et al., 1990). Как правило, имеются парциальные судороги и, как правило, инфантильные спазмы. У большинства пациентов присутствует некоторая степень микроцефалии, обычно легкой. При нехромосомной патологии дисморфизм не выражен, хотя лоб узкий и часто присутствует ретрогнатизм. Прогноз неблагоприятный, с ограниченной выживаемостью.
Некоторые случаи мутации LIS1 могут быть в большей степени связаны с субкортикальными групповыми гетеротопиями, нежели чем с лиссэнцефалией (Gleeson et al., 2000).
Диагноз типа I лиссэнцефалии стал возможным при помощи современных методов нейровизуализации. КТ и МРТ демонстрируют характерный внешний вид широкой кортикальной пластинки, с несколькими присутствующими или отсутствующими извилинами, отделенными от гиподенсивного белого вещества слегка волнистой или почти прямолинейной границей. Слоистость коры может быть выявлена при КТ или МРТ с высокой степенью разрешения. Патологические изменения обычно доминируют в задней части коры, в то время как несколько изгибов можно обнаружить спереди. При ультрасонографии уже с 18,5-25 недели определяется гладкость коры плода или новорожденного (Toi et al., 2004). МРТ дает более точные результаты (Ghai et al., 2006).
На ЭЭГ в большинстве случаев можно увидеть высокоамплитудную быструю активность альфа и бета частот, чередующихся даже на той же записи с высокоамплитудными дельта или тета медленными ритмами, которые могут имитировать медленные комплексы спайк-волн или гипсаритмию (de Rijk-van Andel et al., 1992, Quirk et al., 1993, Mori et al., 1994).
Дифференциальную диагностику проводят с другими состояниями, при которых имеется утолщение коры и нарушение послойного строения. Пахигирия в результате мутации LIS1 считается лишь легкой степенью лиссэнцефалии, не имеющей отношения к дифференциальному диагнозу. Определенные нарушения развития плода, особенно цитомегаловирусная инфекция, по-видимому, могут вызывать развитие пахигирии, гистологически связанной с полимикрогирией. Перивентрикулярная кальцификация может сопровождаться патологией формирования извилин мозга. В таких случаях микроскладки могут сливаться и походить на пахигирию.
Пренатальный диагноз не представляется возможным на поздних сроках беременности с помощью ультрасонографии, поскольку в это время только появляются третичные борозды (Toi et al., 2004). Исследования ДНК могут выявить мутировавший или отсутствующий ген LIS1. Для определения риска рецидива при поиске ламинарных гетеропий необходимы хромосомный анализ и МРТ родителей (особенно матерей).

Классическая (тип I) лиссэнцефалия.
Т1-взвешенная аксиальная МРТ: (мутация LIS I) толстая корковая лента с гладкой поверхностью и прямой, неволнистой границей между серым и белым веществом.
Обратите внимание на присутствие нескольких мелких борозд в лобной области и полное отсутствие борозд сзади,
отсутствие оперкуляции с широко открытой сильвиевой бороздой и слоистость коры со слабой границей между гетеротопированными и полностью мигрировавшими нейронами.
• Субкортикальная ламинарная гетеротопия и лиссэнцефалия в результате мутации DCX гена. Ленточные гетеротопии (Barkovich et al., 1994, Franzoni et al., 1995) или «двойная кора» (Livingston и Aicardi, 1990, Palmini et al., 1991) являются результатом нарушенной миграции, при которой поверхностная кора, внешне нормальная или с отклонениями в извилинах, отделена тонким слоем белого вещества от полосы серого вещества. Граница между серым веществом и подлежащим белым веществом ровная как при агирии-пахигирии. Пациенты с этой аномалией часто страдают судорогами, которые могут иметь очаговый или генерализованный характер, иногда в форме синдрома Ленокса-Гасто и аномальной ЭЭГ (Hashimoto et al., 1993, Parmeggiani et al., 1994).
Нарушения умственного развития значительно варьируют, некоторые пациенты развиваются нормально (Livingston и Aicardi, 1990, Ianetti et al., 1993). Barkovich et al. (1994) при детальном изучении 27 случаев обнаружили значительную корреляцию между интеллектуальным уровнем и толщиной гетеротопической полосы; внешне нормальная кора была связана с лучшим развитием, но, вероятно, этот признак может варьировать. На ЭЭГ было обнаружено, что полоса способна продуцировать пароксизмальную активность и повышенный кровоток, как было продемонстрировано с помощью ОФЭКТ, что указывает на активацию коры.
Это состояние в большинстве случаев обусловлено сцепленной с полом мутацией DCX гена, кодирующего даблкортин (англ, doublecortin) (des Portes et al, 1998, Gleeson et al., 1999). Тем не менее, сходная мутация у мальчиков может привести к классической лиссэнцефалии (Pilz et al., 1998). Мутация очень разнообразно выражается у женщин и даже у некоторых мужчин (Cardoso et al. 2000, Gleeson 2000) и поэтому трудно распознается. Поэтому генетическое консультирование семьи, где родился мальчик с лиссэнцефалией, должно включать тщательный поиск ламинарной гетеротопии на МРТ и, если необходимо, мутации DCX у матери и сестер. Известны семьи, где пораженная мать рождала мальчиков с лиссэнцефалией и девочек с ламинарными гетеротопиями (Pinard et al., 1994). Редкие случаи ламинарной гетеротопии связаны с миссенс-мутацией LIS1 и с более умеренным фенотипом (Leventer et al., 2001).
При визуализации, тяжесть экспрессии у девочек варьирует от широких субкортикальных полос, иногда покрытых патологической корой, до с трудом обнаруживаемых тонких полос, которые видны только под ограниченными участками коры. Односторонние и частичные линейные гетеротопии иногда сложно распознать, для выявления могут потребоваться специальные срезы и изменение формата MPT (Gallucci et al., 1991). У мальчиков картина классической лиссэнцефалии такая же, как при LIS1. Однако передние отделы коры имеют более гладкую поверхность в сравнении с задними, в отличие от происходящего при мутации LIS1.
Эпилепсия, связанная с ламинарными гетеротопиями, может поддаваться медикаментозному лечению, но бывает и устойчивой. Хирургическое лечение оказалось неэффективным.
Синдром Барайтсера-Уинтера включает дисмор-фические признаки и пороки развития мозга в виде классической лиссэнцефалии или субкортикальных ламинарных гетеротопий (Rossi et al., 2003).
• Пахигирия. Этот тип представляет менее тяжелую форму спектра лиссэнцефалии и, вероятно, возникает в результате тех же механизмов. Однако эта форма гетерогенна и может входить в состав разных синдромов. Клинически пахигирия представлена различными схожими симптомами, но с меньшей тяжестью. На МРТ выявляют утолщение коры и линейное разделение между корой и белым веществом.

(а) Лиссэнцефалия-пахигирия у двухлетней девочки: ЭЭГ указывает на типичные быстрые ритмы с альфа и выше частотой.
(б| Синдром Миллера-Дикера у 14-недельной девочки: ритмическая активность различных частот, но в основном в тета-диапазоне.
(в) Синдром Миллера-Дикера у двухлетнего мальчика: хотя присутствует некоторая избыточная тета-альфа активность, в записи преобладают повторяющиеся вспышки острых волн, достигающих 500-600 μВт.
• Другие формы и синдромы лиссэнцефалии. Распознание некоторых менее распространенных вариантов лиссэнцефалии не менее важно из-за разницы генетических и прогностических последствий (Hennekam и Barth, 2003, Raoul et al., 2003).
Микролиссэнцефалия состоит из крайне выраженной врожденной микроцефалии и агирии или пахигирии с широкой корой. Описано по меньшей мере, пять или шесть типов, передающихся по рецессивному типу, с различной степенью утолщения кортикального слоя, расположением имеющихся борозд и наличием сопутствующих пороков, таких как гипоплазия мозжечка, стволовая атрофия и увеличение желудочков (Ross et al., 2002, Dobyns и Leventer, 2003, Sztriha et al., 2004). Некоторые авторы (Dobyns и Barkovich, 1999) выделили эти случаи из «олигирической микроцефалии» (Hanefeld, 1999), которую они расценивают скорее как форму первичной микроцефалии, нежели форму расстройства миграции. Один из этих синдромов может быть связан с мутацией рилин гена (Hong et al., 2000, Crino, 2001).
Лиссэнцефалия с гипоплазией мозжечка является отдаленным проявлением микроцефалии с рудиментарной двуслойной корой мозга и тяжелой гипоплазией мозжечка (Ross et al., 2001, Sztriah et al., 2005). Вероятно, с рецессивным наследованием.
Лиссэнцефалия с гипоплазией мозолистого тела генетически гетерогенна. Некоторые случаи могут входить в группу мутации LIS1 или микролиссэнцефалии.
Х-сцепленная лиссэнцефалия с аномалией гениталий (XLAG) — врожденный порок с микроцефалией, тяжелой задержкой развития, тенденцией к гипотермии, отсутствием мозолистого тела и множественными аномалиями мозга (Berry-Kravis и Israel, 1994, Dobyns et al., 1999). Более вероятна гипоплазия гениталий, чем агенезия. XLAG развивается в результате мутации гомеобоксного гена ARX на хромосоме Х33.2 (Uyanik et al., 2003), на которой другие мутации также могут быть причиной некоторых неврологических синдромов (Kato et al. 2004, Suri 2005), включая Х-сцепленную задержку умственного развития (MRX54), агенезию мозолистого тела с патологией гениталий и синдром Партингтона с умственной отсталостью, атаксией и дистонией в зависимости от типа мутации.
Интересно, что лиссэнцефалия с неонатальными судорогами и тяжелыми аномалиями развития нервной системы, как было выявлено, связана с отсутствием глутамина.

Субкортикальная групповая гетеротопия («двойная кора»):
(а) Аксиальный срез МРТ: широкие, непрерывные группы с таким же сигналом как от коры.
(б) Коронарный срез: в этом же случае имеется расширение желудочков преимущественно спереди.
(в, г) MPT, Т1-взвешенная последовательность — (в) аксиальный срез, (г) сагиттальный срез -тонкий слой белого вещества, лежащий между истинной корой и тонкой линейной гетеротопией серого вещества (стрелки).
Аномалии развития головного мозга – синдром «двойной коры»
Большинство врожденных пороков центральной нервной системы представляют собой мультифакториальную патологию эмбрионального периода развития. Неврологическая симптоматика аномалий развития головного мозга зависит от их локализации и объема поражения. Кроме того, на степень неврологических расстройств может повлиять специфика патологической архитектоники тканей головного мозга и их соотношения между собой. Клиническая симптоматика этих аномалий малоспецифична. К наиболее частым неврологическим симптомам относят центральные парезы, эпилептические приступы, а также задержку психического и моторного развития различной степени выраженности. Среди наиболее часто встречаемых аномалий развития головного мозга выделяют кортикальные дисплазии, которые включают: фокальную кортикальную дисплазию, региональную и диффузную пахигирию, унилатеральную гемимегалэнцефалию, голопрозэнцефалию, шизэнцефалию, нейронные гетеротопии.
Фокальная корковая дисплазия – это очаговое нарушение нейронной миграции и дифференцировки. Выделяют несколько типов фокальных корковых дисплазий: тип 1, при котором нарушается корковая нейрональная организация при сохранности пирамидального рисунка коры, и тип 2, при котором имеет место выраженная дезорганизация с потерей пирамидального рисунка, при этом наблюдаются гигантские (баллонные) клетки 3. Основной локализацией фокальных корковых дисплазий является височная доля, наиболее эпилептогенная структура головного мозга. Агирия (лиссэнцефалия) – нарушение нейрональной дифференциации с уменьшением числа извилин вплоть до гладкого мозга. Характерен симптомокомплекс: микроцефалия, диффузная мышечная гипотония, эпилептические спазмы. Регионарная корковая дисплазия чаще представлена врожденным перисильвиарным синдромом. Суть нейроморфологических изменений заключается в билатеральной оперкулярной дисгирии. В клинической картине доминируют эпилептические приступы, псевдобульбарный и пирамидный синдром. Унилатеральная гемимегалэнцефалия – увеличение размеров одной доли или ее части вследствие избыточной пролиферации нейронов. Проявляется эпилептическими приступами, контрлатеральным гемипарезом [1, 2]. Голопрозэнцефалия – порок развития, при котором мозг остается нерасщепленным, часто сочетается с аномалиями лицевого скелета и приводит к летальному исходу в ранний постнатальный период. Шизэнцефалия проявляется «расщелинами» мозга, преимущественно в височной доле. В неврологическом статусе чаще наблюдаются резистентный эпилептический синдром, двигательные расстройства 1. Нейронные гетеротопии – нарушения нейрональной миграции на 35-й неделе гестации с образованием эктопированных участков нодулярной или ламинарной формы.
Согласно данным литературы, нейронные гетеротопии ответственны за 5-25% случаев эпилепсии у детей [1].
Наиболее показательным вариантом аномалии развития головного мозга является вариант ламинарной гетеротопии, когда слои гетеротипированных нейронов располагаются в глубоких и субкортикальных отделах головного мозга, известный как синдром «двойной коры».
Синдром «двойной коры» – это редкая, генетически обусловленная аномалия развития центральной нервной системы. Ее возникновение связано с мутацией гена даблкортина, локализованного в хромосоме Xg22, которая приводит к формированию ламинарной (ленточной) подкорковой гетеротопии нейронов. Вследствие подобного нарушения миграционных процессов создается иллюзия дублирования коры – «двойная кора» 3. Синдром впервые описан H. Jakob в 1936 г. и в дальнейшем выявлен S. Ricci и A Palmini у больных с эпилептическими синдромами [6]. В клинической картине синдрома наиболее часто наблюдаются задержка психомоторного развития, терапевтически резистентная эпилепсия с преобладанием парциальных/астатических приступов и дебютом припадков преимущественно после 5 лет, четкие очаговые изменения на электроэнцефалограмме (ЭЭГ), могут также встречаться инфантильные спазмы в анамнезе. Лечение этого синдрома симптоматическое, основой которого является противоэпилептическая терапия [1, 6].
Ниже приведен случай, отвечающий основным диагностическим критериям синдрома «двойной коры».
Клинический случай
Анамнез жизни и заболевания
Пациентка Г., 1995 г. р., родилась от четвертой беременности (1 – самопроизвольный аборт в ранние сроки, 2 – роды, здоровая дочь, 20 лет, 3 – медицинский аборт). Беременность протекала с угрозой прерывания на ранних сроках. Роды были срочные, физиологические. Масса при рождении составила 3200 кг, оценка по шкале Апгар – 8/8 баллов. Раннее моторное и речевое развитие проходило с некоторой темповой задержкой. В 5-летнем возрасте у нее появились серийные приступы «остановки» взгляда с замиранием, затем добавились фокальный компонент с тонической девиацией глаз влево и тонико-клонические подергивания в левой руке, далее – вторично генерализованные пароксизмы. Была проведена терапия фенобарбиталом и вальпроевой кислотой. В возрасте 10 лет у пациентки появились атонические, затем – аутомоторные приступы, к терапии добавили ламотриджин. Отмечено нарастание двигательных нарушений с формированием тетрапареза и когнитивных нарушений.
Состояние при поступлении
При поступлении в стационар состояние пациентки по основному заболеванию классифицировалось как тяжелое. В неврологическом статусе: правая глазная щель была больше левой, зрачки равны, отмечено вертикальное косоглазие слева, сглажена правая носогубная складка, наблюдалась девиация языка и язычка влево. Мышечный тонус в конечностях дистоничен, без разницы сторон, движения в конечностях ограниченные, мышечная сила снижена в проксимальных отделах конечностей, сухожильные рефлексы равномерно оживлены, равны, патологические стопные знаки отмечаются с двух сторон, в пробе Ромберга отклонение назад и в стороны. Пальценосовую пробу выполняет с мимопопаданием. У больной имеет место лишний вес. Словарный запас и интеллект снижены.
Результаты обследования
Согласно данным нейропсихологического исследования, коэффициент интеллекта (IQ) пациентки соответствовал 62 баллам.
У больной был проведен ЭЭГ-мониторинг в течение 24 часов (аппарат электроэнцефалограф-регистратор «Энцефалан-ЭЭГр-19/86», производство «Медиком-мтд» г. Таганрог, Россия): во время бодрствования и ночного сна в лобных отведениях регистрировалась эпилептиформная активность в виде комплексов острая волна – медленная волна с тенденцией к генерализации (рис. 1).
 |
Кроме того, была проведена магнитно-резонансная томография головного мозга (аппарат Hitachi Airis Mate 0,2 Тесла), согласно которой на аксиальных срезах определялись билатеральные лентовидные зоны, соответствующие серому веществу головного мозга, расположенные преимущественно субкортикально. Изгибы гетеротопированных слоев повторяли основную складчатость кортикальной поверхности. На коронарных срезах подтверждалось субкортикальное расположение гетеротопированных зон. В коре видимых диспластических изменений не отмечено. Таким образом, можно утверждать о наличии у пациентки МР-признаков билатеральной ламинарной гетеротопии серого вещества, что характерно для синдрома «двойной коры» (рис. 2).
 |
Обоснование диагноза и лечения
Таким образом, у больной отмечался ранний дебют эпипароксизмов со специфической динамикой и наслоением пароксизмов: фокальные – вторичная генерализация – астатические – аутомоторные пароксизмы, нарастающий когнитивный и неврологический дефицит, преобладание фокальной эпилептической активности на ЭЭГ и, наконец, наиболее значимый диагностический критерий – МР-признаки ламинарной гетеротопии серого вещества. В ходе обследования был выставлен диагноз: «аномалия развития центральной нервной системы: билатеральная ламинарная гетеротопия серого вещества головного мозга – синдром «двойной коры», эпилептическая энцефалопатия Леннокса – Гасто».
Пациентке была назначена противоэпилептическая терапия двумя препаратами – леветирацитамом в дозе 2000 мг/сут и ламотриджином по 200 мг/сут.
Катамнез в течение 6 месяцев показал купирование атонических приступов, но сохранение фокальных и аутомоторных. В перспективе возможна модификация противопилепической терапии: зонисамид, этосуксимид, лакосамид. Также обсуждается вопрос нейрохирургической коррекции для уменьшения количества пароксизмов.
Выводы
Рассмотренный случай подчеркивает необходимость придерживаться ряда облигатных принципов, ставших в ведущих эпилептологических центрах рутинными в повседневной практике врача-эпилептолога. К ним относятся такие принципы, как корректная синдромологическая диагностика пароксизмов, пролонгированный ЭЭГ-видеомониторинг, магнитно-резонансная томография высокой разрешимости по протоколу эпилептологического сканирования, генетическое типирование, что позволяет своевременно и точно диагностировать искомую патологию.
Использование магнитно-резонансной томографии является принципиально важным диагностическим инструментом для уточнения этиопатогенеза эпилепсии даже при наличии идиопатической ее формы. Трудно оценить всю значимость своевременного этиологического диагноза для выбора рациональной терапии, определения прогноза и семейного консультирования.
Литература
- Алиханов А.А. Нейрорадиологическая модель различных вариантов нарушения нейронной миграции // Журнал неврологии и психиатрии. – 2004. – № 10. – С. 81-85.
- Шестова Е.П., Евтушенко С.К., Соловьева Е.М., Душацкая А.В. Аномалии головного мозга (миграционные нарушения) у детей: клинико-радиологические проявления //Международный неврологический журнал. – 2005. – № 4 (4). – С. 30-36.
- Коновалов А.Н., Корниенко В.Н., Озерова В.И., Пронин И.Н. Нейрорентгенология детского возраста. – М.: Андор, 2001. – 456 с.
- Cohen M.M., Jr. The Child with Multiple Birth Defects / Second edition. – New York: Oxford University Press, 1997. – 267 p.
- Neil G. Epilepsy and Disorders of Neuronal Migration. I Introduction // Developmental Medicine and Child Neurology. – 1996. – V. 38. – Р. 1053-1057.
- Palmini A., Rim E-H., Da Costa J.C. Evidence for Focal accentuation if cortical dysfunction/excitability in the «Double cortex» syndrome // Epilepsy. – V. 38 (Suppl 3). – P. 6.
1 Детское клиническое территориальное медицинское объединение, г. Макеевка.
2 2 ООО « Медицинская лучевая диагностика», г. Макеевка.
Аномалии развития центральной нервной системы , страница 8
а - Т1-ВИ, сагиттальная плоскость. Агирия затылочной доли. Извилины теменной доли утолщены, широкие.
б - IR ИП, аксиальная плоскость. Толщина коры увеличена, желудочки мозга расширены.
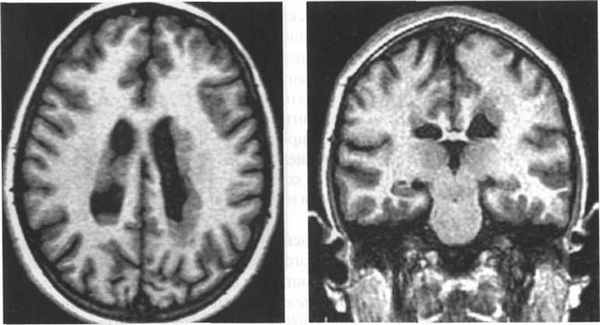
Рис. 3.19. Перивентрикулярная гетеротопия. МРТ. а - IR ИП, аксиальная плоскость; б - IR ИП, корональная плоскость.
Множественные узлы гетеротопии располагаются вдоль стенок боковых желудочков.
Различают следующие формы гетеротопии: перивентрикулярную узловую, перивентрикулярную и субкортикальную как с изменением, так и без изменения строения коры, гигантскую, сочетающуюся с кортикальной дисплазией, и лентовидную.
Перивентрикулярная узловая гетеротопия характеризуется четко очерченными узлами, расположенными вдоль стенки желудочка мозга. Узлы могут быть как одиночными, так и множественными и обычно вдаются в полость желудочка (рис. 3.19).
Перивентрикулярная и субкортикальная гетеротопия как с изменением, так и без изменения строения коры проявляется узловой перивентрикулярной гетеротопией и скоплением серого вещества в субкортикальных отделах. Поражение в большинстве случаев одностороннее. Субкортикальное скопление серого вещества может приводить к локальной деформации борозд и утолщению коры (рис. 3.20).
Гигантская форма гетеротопии с изменением строения коры - большое по протяженности скопление серого вещества, занимающее большую часть гемисферы, от стенки желудочка до поверхности коры, приводящее к деформации кортикальной поверхности мозга. При данной форме гетеротопии скопления серого вещества в виде отдельных узлов не наблюдается. Гигантскую форму гетеротопии, вследствие большого размера зоны поражения, необходимо дифференцировать с патологическими образованиями. При гетеротопии, в отличие от опухолей, не определяются перифокальный отек, смещение срединных структур, нет усиления сигнала после введения контрастирующего вещества.


Рис. 3.20. Перивентрикулярно-субкортикальная гетеротопия. МРТ.
а - IR ИП, аксиальная плоскость. Узлы гетеротопии располагаются вдоль стенки левого бокового желудочка и в субкортикальных отделах белого вещества. Между субкортикальными узлами сохраняются прослойки белого вещества. Поверхность коры деформирована.
б - Т2-ВИ, корональная плоскость. Субэпендимальные узлы вдаются в полость левого бокового желудочка, что делает его контуры волнистыми.
Лентовидная гетеротопия, или синдром двойной коры, проявляется четко очерченным лентовидным слоем нейронов, отделенным от коры полосой белого вещества. Диагностировать данную патологию можно только по данным МРТ. При этом на изображениях выявляется ровная, четко очерченная полоса серого вещества, расположенная параллельно боковому желудочку и отделенная от коры и стенки желудочка слоем серого вещества. Кора мозга может быть неизмененной либо может быть изменена от умеренно выраженной пахигирии до полной агирии (рис. 3.21). В белом веществе на Т2-ВИ могут определяться очаги гиперинтенсивного сигнала. Лентовидную гетеротопию достаточно сложно дифференцировать с лиссэнцефалией: они, вероятно, представляют собой различные степени одного общего процесса нарушения миграции нейронов. В отличие от лиссэнцефалии, при лентовидной гетеротопии изменения коры выражены меньше.


Рис. 3.21. Лентовидная гетеротопия. МРТ.
а - IR ИП, аксиальная плоскость; б - Т2-ВИ, аксиальная плоскость.
Полоса гетеротопированного серого вещества отделена
слоем белого вещества от коры и желудочков мозга.


Рис. 3.22. Двусторонняя открытая шизэнцефалия. МРТ.
а - Т2-ВИ, аксиальная плоскость; б - Т1-ВИ, корональная плоскость.

Рис. 3.23. Открытая шизэнцефалия правой лобной доли. МРТ.
а - IR ИП, аксиальная плоскость.
Края расщелины, располагающейся в правой лобной доле, представлены диспластичным серым веществом. Полость расщелины заполнена ликвором. В левой гемисфере определяется изменение хода борозд и утолщение коры.
б - Т1-ВИ, корональная плоскость.
В лобной доле выявлена расщелина сложной формы с образованием нескольких небольших слепо заканчивающихся ответвлений. Прилежащее субарахноидальное пространство и передний рог бокового желудочка расширены.
Шизэнцефалия представляет собой вариант кортикальной дисплазии, когда определяется расщелина, проходящая через все полушарие головного мозга - от бокового желудочка до кортикальной поверхности. Клинические симптомы зависят от степени выраженности изменений и проявляются судорогами, гемипарезом, отставанием в развитии. Наиболее часто расщелина локализуется в пре-и постцентральной извилине и может быть как односторонней, так и двухсторонней (рис. 3.22). В большинстве случаев при унилатеральной шизэнцефалии в контралатеральной гемисфере выявляются другие виды кортикальных дисп-лазий (пахигирия, полимикрогирия) (рис. 3.23). В области расщелины прослеживаются крупные сосуды. Серое вещество, покрывающее расщелину, диспластичное, утолщенное, имеет неровную внутреннюю и наружную поверхность.
Читайте также: