Болезнь Ниманна-Пика
Обновлено: 25.04.2024
Болезнь Ниманна-Пика является сфинголипидозом Сфинголипидоз Лизосомальные ферменты разрушают макромолекулы либо самой клетки (например, когда перерабатываются структурные компоненты клетки), либо захваченные извне. Унаследованные дефекты или недостатки. Прочитайте дополнительные сведения , наследственным нарушением обмена веществ, которое вызвано недостаточной активностью сфингомиелиназы, что приводит к накоплению сфингомиелина (церамидного фосфорилхолина) в ретикулоэндотелиальных клетках. Диагноз ставят на основании анализа ДНК и/или анализа ферментов лейкоцитов. Вариантами лечения могут быть трансплантация костного мозга, трансплантации стволовых клеток и ферментозамещающая терапия.
Для получения дополнительной информации см. таблицу Некоторые сфинголипидозы Некоторые сфинголипидозы и таблицу Прочие липидозы Другие липидозы .
Наследование болезни Ниман-Пика - аутосомно-рецессивные Аутосомно-рецессивные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения и чаще всего возникает у евреев ашкенази; существует 2 типа – А и В. Тип С болезни Ниманна Другие липидозы – Пика связан с дефектами ферментов, вовлекающих аномальное хранение холестерина.
Дети с типом А имеют показатели активности сфингомиелиназы 5% от нормы. Заболевание характеризуется гепатоспленомегалией, задержкой развития и быстро прогрессирующей нейродегенерацией. Смерть наступает в возрасте 2 или 3 лет.
У пациентов с типом B активность сфингомиелиназы составляет 5–10% от нормы. Тип B имеет более вариативные клинические проявления, чем тип А. Могут развиться гепатоспленомегалия и лимфаденопатия. Панцитопения довольно распространена. Большинство пациентов с типом B практически не имеют неврологических поражений и доживают до взрослого возраста, они могут быть клинически неотличимы от лиц с болезнью Гоше типа I Болезнь Гоше Болезнь Гоше является сфинголипидозом – наследственным нарушением метаболизма, которое возникает в результате дефицита глюкоцереброзидазы и приводит к накоплению глюкоцереброзида и родственных. Прочитайте дополнительные сведения . В тяжелых случаях типа B прогрессирующие легочные инфильтраты вызывают основные осложнения.
Диагностика болезни Ниманна-Пика
Исследование сфингомиелиназы лейкоцитов
Оба типа обычно подозревают по истории болезни и результатам исследований, в первую очередь гепатоспленомегалии. Диагноз болезнь Ниманна-Пика может быть подтвержден на основании анализа ДНК и/или анализа сфингомиелиназы лейкоцитов, и это может быть сделано пренатально с помощью амниоцентеза или биопсии хориона. Анализ ДНК может быть также проведен для определения носителей. (Также см. Исследования при подозрении наследственных нарушений обмена веществ Начальное тестирование Большинство наследственных нарушений метаболизма (врожденные нарушения обмена веществ) являются редкими, и, следовательно, для их диагностики необходим более высокий индекс подозрения. Своевременная. Прочитайте дополнительные сведения ).
Лечение болезни Ниманна-Пика
Возможна трансплантация костного мозга, стволовых клеток и заместительная терапия ферментом
Трансплантация костного мозга, трансплантации стволовых клеток, и заместительная терапия ферментом изучаются в качестве потенциальных вариантов лечения.
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Online Mendelian Inheritance in Man® (OMIM®) database: полная информация о генах и их молекулярной и хромосомной локализации
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Болезнь Ниманна-Пика
ФГБУ «Национальный медицинский исследовательский центр кардиологии» Минздрава России, Москва, Россия
Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
ГУ Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
кафедра нервных болезней педиатрического факультета Российского государственного медицинского университета, Москва
ГБУЗ МО «Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского», Москва, Россия
"Алтайский государственный медицинский университет" Минздрава России, Барнаул
Болезнь Ниманна—Пика типа С у ребенка
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2017;117(11‑2): 62‑66
ФГБУ «Национальный медицинский исследовательский центр кардиологии» Минздрава России, Москва, Россия
В статье приводится случай болезни Ниманна—Пика типа С (БНП-С) — орфанного наследственного аутосомно-рецессивного нейродегенеративного заболевания, принадлежащего к группе лизосомных болезней накопления, у пациентки 11 лет с поздней младенческой формой болезни. Сочетание задержки психомоторного развития, полиморфной неврологической симптоматики и соматических изменений в виде изолированной спленомегалии позволили предположить у пациентки БНП-С. Ей была проведена ДНК-диагностика методом прямого автоматического секвенирования, которая подтвердила диагноз. Больной было назначено лечение миглустатом.
ФГБУ «Национальный медицинский исследовательский центр кардиологии» Минздрава России, Москва, Россия
Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
ГУ Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского
кафедра нервных болезней педиатрического факультета Российского государственного медицинского университета, Москва
ГБУЗ МО «Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского», Москва, Россия
"Алтайский государственный медицинский университет" Минздрава России, Барнаул
Болезнь Ниманна—Пика типа С (БНП-С) — орфанное наследственное аутосомно-рецессивное нейродегенеративное заболевание, относящееся к классу лизосомных болезней накопления. Распространенность БНП-С в мире составляет около 1:120 000—1:150 000 живых новорожденных. Болезни свойственны разнообразные неврологические проявления, включая неуклюжесть, атаксию, дизартрию, дисфагию и снижение когнитивных функций. Болезнь характеризуется вариабельной скоростью прогрессирования. В терминальной фазе заболевания, пациенты, как правило, не способны самостоятельно двигаться и проглатывать пищу, у них грубо нарушены когнитивные функции. БНП-С является тяжелым инвалидизирующим заболеванием и оказывает эмоциональную и экономическую нагрузку на пациентов, их семью и общество [1, 2].
Болезнь Ниманна—Пика в 1961 г. была разделена А. Crocker на 4 типа (А, В, С, D). Ранее существовало мнение, что все фенотипы обусловлены изменениями в гене сфингомиелиназы. Гены NPC1 и NPC2 (мутации в которых вызывают БНП-С) были картированы в конце 90-х годов. В 95% случаев БНП-С вызвана мутациями в гене NPC1 (локус 18q11-q12), ответственного за синтез транспортного стирол-чувствительного белка клеточной мембраны. Примерно 5% случаев БНП 2-го типа обусловлена мутациями в гене NPC2 (локус 14q24), отвечающего за синтез лизосомального белка-транспортера холестерина. Дисбаланс метаболизма липидов и холестерина приводит к накоплению сфингомиелина и других фосфолипидов и холестерина в лизосомах органов-мишеней (ЦНС, ретикуло-эндотелиальная система) [3—6].
Клинические симптомы БНП-С разнообразны, они представляют собой сочетание неврологических, висцеральных, психиатрических и системных признаков, не являющихся специфичными для заболевания, манифестируют в разном возрасте и характеризуются различной скоростью прогрессирования, что представляет собой значительные сложности в диагностике [7—9]. В соответствии с возрастом дебюта было выделено несколько клинических форм БНП-С: ранние формы (неонатальная — манифестация до 3 мес; ранняя младенческая — от 3 мес до 2 лет; поздняя младенческая — от 2 до 6 лет), ювенильная форма (6—12 лет), взрослая форма (12 лет и старше) [2]. Неонатальная форма БНП-С характеризуется тяжелым течением, сопровождается внутриутробной водянкой плода, часто в первые недели жизни присоединяется холестатическая желтуха в сочетании с гепатоспленомегалией. Неврологические симптомы представлены гипотонией мышц и нарушением психомоторного развития. Течение заболевания крайне тяжелое, нередко с летальным исходом к 1-му году жизни. Для более поздних форм свойственно сочетание неврологических, психических и соматических нарушений, среди которых изолированная «деликатная» спленомегалия, как правило, случайно выявляемая при УЗИ, вертикальный супрануклеарный паралич взора, атаксия конечностей, дизартрия, дисфагия, пирамидная симптоматика (оживление глубоких и поверхностных рефлексов), периферическая полинейропатия, экстрапирамидные нарушения (тремор, дистонические гиперкинезы и др.), эпилептические приступы, геластическая катаплексия (приступы мышечной гипотонии, провоцируемые сильными эмоциональными реакциями) [1, 7]. Ниже приводим наблюдение.
Пациентка П., 11 лет, младшая дочь в неродственном браке здоровых родителей европейского происхождения. Сибс (брат 17 лет) клинически здоров. Родилась от 3-й беременности, протекавшей с отслойкой плаценты в 12 нед, хронической внутриутробной гипоксией плода. Роды на 41-й неделе, самопроизвольные, масса тела 3910 г, длина 51 см, оценка по шкале АПГАР — 8/9 баллов. Раннее развитие: самостоятельно пошла в 1 год. Фразовая речь с 2 лет. С 3—4 лет появились неуклюжесть, тремор в руках. В 7 лет возникли трудности в обучении, атаксия конечностей. Была проконсультирована в детском неврологическом центре с заключением: «синдромальная форма задержки развития, тремор».
В 7 лет была проконсультирована в Институте коррекционной педагогики, диагноз: «выраженная задержка психического развития; сенсомоторная алалия; дизартрия; экстрапирамидный синдром».
Кроме того, была направлена на консультацию врача-генетика с целью проведения дифференциальной диагностики с наследственными атаксиями и дегенеративными заболеваниями ЦНС. Был выполнен хромосомный микроматричный анализ, патогенного хромосомного дисбаланса выявлено не было, патогенные микроделеции и микродупликации размером более 50 000 п.н. не были обнаружены. Участки потери гетерозиготности, содержащие гены, связанные с феноменом импритинга, — отсутствовали. Протяженные участки потери гетерозиготности (>3 000 000 п.н.) — 0,11% (общепопуляционный уровень).
В течение последующего времени наблюдалось прогрессирующее снижение интеллекта, нарастание нарушений координации.
В 9 лет была проконсультирована клиническим психологом. Отмечены значительное снижение общего интеллектуального развития, низкий запас знаний и представлений об окружающей среде, бедный словарный запас, снижение объема внимания. В этом же возрасте развился первый приступ эпилепсии. Наблюдалась с диагнозом криптогенная эпилепсия, проводимая терапия была неэффективна. В возрасте 11 лет появились признаки дисфагии, патологическая поза левой ноги (ротация голеностопного сустава кнутри).
При консультации в Медико-генетическом научном центре в возрасте 11 лет была заподозрена БНП-С. При неврологическом осмотре соматический статус был без существенных особенностей. Называла свое имя, возраст с подсказкой. Периодически стереотипно подкашливала. Речь растянутая. Легкая гиперактивность. Поведение корригировалось после нескольких повторений. Глазные щели и зрачки равной величины, фотореакции живые, ограничение самопроизвольных движений глазных яблок вверх и при слежении за молоточком. Нистагма нет. Асимметрии лицевой мускулатуры нет. Язык по средней линии. Мышечный тонус был умеренно снижен в проксимальных отделах, дистальных отделах — дистоничный, в ногах — клонусы. Атетоидная установка кистей рук. Гиперкинезы в руках. Сухожильная гиперрефлексия. Грубый интенционный тремор. Поперхивалась во время еды.
Данные лабораторных и инструментальных исследований
Биохимическое исследование плазмы и сухих пятен крови пациентки: концентрация триола 33,4 нг/мл (норма 2,0—50 нг/мг), концентрация 7-кетохолестерина — 83,4 нг/мл (норма 10—75 нг/мг), активность хитотриозидазы — 51 нмоль/ч/мл (норма 2,5—100 нмоль/ч/мл).
Молекулярно-генетическое исследование: проведен полный анализ гена NPC1, методом прямого автоматического секвенирования проанализированы кодирующие экзоны гена 1—25 и прилегающие интронные участки. Выявлена описанная в базах данных мутация с.3070C>T (p.Pro1007Ala) в 20 экзоне гена в гетерозиготном состоянии и неописанная замена с.3148G>А (p.Asp1050Asn) в 21 экзоне гена в гетерозиготном состоянии. Проведен анализ патогенности замены c.3148G>A: базы MutationTaster, PolyPhen-2, PROVEAN, SIFT — патогенная замена. Проведено молекулярно-генетическое исследование крови родителей и брата пробанда: у матери мутация с.3148G>А в гетерозиготном состоянии (носитель), у отца — с.3070С>G в гетерозиготном состоянии (носитель), у брата — с.3070С>G в гетерозиготном состоянии (носитель).
Ночной видео-ЭЭГ-мониторинг: основная активность сформирована в пределах возрастной нормы, дезорганизована низкочастотными волнами (рис. 1-3). Рис. 1. Ночной видео-ЭЭГ-мониторинг больной от 04.11.16—05.11.16 (бодрствование). Рис. 2. Ночной видео-ЭЭГ-мониторинг больной от 04.11.16—05.11.16 (бодрствование). Рис. 3. Ночной видео-ЭЭГ-мониторинг больной от 04.11.16—05.11.16 (сон). В бодрствовании частотные характеристики основной активности в затылочных отделах полушарий головного мозга в пределах возрастной нормы — 8—8,5 Гц. В бодрствовании и во сне регистрируется преходящее региональное тета-, дельта-замедление в правой височно-теменно-затылочной области, реже в левой лобно-центрально-височной области. Устойчиво регистрируются мультирегиональные эпилептиформные изменения в виде комплексов пик-медленная волна, острая-медленная волна в левой затылочной области с тенденцией к латерализации на все левое полушарие; в правой лобно-центрально-височной области, независимо в левой задне-лобно-центральной области с распространением на задние отделы соименного полушария и дистантным распространением на гомологичные отделы противоположного полушария, а также короткие диффузные низкосинхронизированные разряды комплексов острая—медленная волна чаще с акцентом в правом полушарии, длительностью не более 0,1—0,5 с. Представленность диффузных эпилептиформных разрядов уменьшается во сне. В целом в бодрствовании и во сне индекс представленности эпилептиформной активности достигает 35—40% на продолженных эпохах записи.
Ультразвуковое исследование внутренних органов — умеренно выраженная спленомегалия.
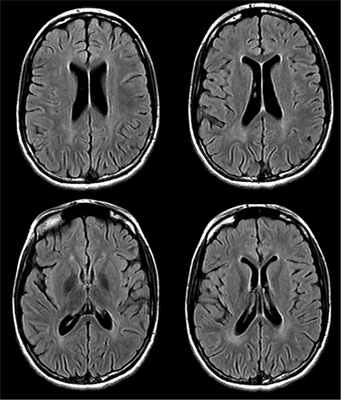
На МРТ головного мозга, выполненной в возрасте 11 лет, выявляется диффузная церебральная атрофия, задержка миелинизации перивентрикулярного белого вещества задних отделов больших полушарий, отмечается отрицательная динамика по сравнению с предыдущими исследованиями (рис. 4). Рис. 4. МРТ головного мозга от 30.09.16.
Клинический диагноз: БНП-С (гликосфинголипидоз, лизосомная болезнь накопления липидов), поздняя младенческая форма, аутосомно-рецессивный тип наследования. Симптоматическая мультифокальная эпилепсия с ороалиментарными автоматизмами, вторично-генерализованными судорожными приступами, фокальными диалептическими приступами.
В данном наблюдении имело место постепенное начало заболевания при отсутствии грубых изменений головного мозга, неэффективность проводимой противоэпилептической терапии, выявленная «деликатная» спленомегалия, вертикальный супрануклеарный паралич взора. Сочетание задержки психомоторного развития, полиморфной неврологической симптоматики и соматических изменений в виде изолированной спленомегалии позволили предположить у пациентки БНП-С.
Сложность диагностики заболевания в более раннем возрасте заключается в трудностях объединения неспецифических клинических проявлений БНП-С, таких как задержка развития, эпилепсия, неуклюжесть, в единый симптомокомплекс одного заболевания.
В помощь практическим врачам для облегчения клинической диагностики БНП-С F. Wijburg и соавт. [10, 11] предложили балльный диагностический индекс вероятности БНП-С. Каждому симптому заболевания присваивается определенное количество баллов в зависимости от уровня риска. При общей сумме баллов
В настоящее время разработана патогенетическая субстрат-редуцирующая терапия для БНП-С. Единственным разрешенным в ряде стран лекарственным препаратом для лечения БНП-С является миглустат. Механизм действия субстрат-редуцирующей терапии — ингибирование фермента глюкозилцерамидсинтазы, отвечающего за первый этап синтеза большинства гликосфинголипидов (уменьшает их накопление в клетке). Своевременная диагностика заболевания и назначение специфической и симптоматической терапии увеличивают продолжительность жизни больных и улучшают ее качество. Пациенты должны получать субстрат-редуцирующую терапию пожизненно. Больной П., представленной в клиническом наблюдении, был назначен специализированный препарат сразу после подтверждения диагноза.
Новые подходы к диагностике болезни Ниманна-Пика типа С
Болезнь Ниманна-Пика типа С (НП-С) представляет собой редкое аутосомно-рецессивное нейродегенеративное заболевание, истинная частота которого недооценивается из-за неспецифичности клинических симптомов. Для болезни НП-C характерен выраженный клинический полиморфизм с различными сроками манифестации заболевания от внутриутробного и неонатального периода до взрослого возраста. В последние годы широкое использование биомаркеров для селективного скрининга и генетического тестирования с использованием панелей генов способствовало повышению доли пациентов, выявленных на ранних этапах развития болезни. В статье описаны наиболее значимые клинические проявления и их комбинации, а также обсуждаются изменения, внесенные в существующие клинические рекомендации по диагностике и лечению болезни НП-С.
Ключевые слова
Об авторах
ФГБУ «Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии им. Кулакова В.И.» Минздрава РФ; ФГАОУ ВО Первый Московский государственный медицинский университет им. И.М.Сеченова Минздрава РФ
Россия
ФГБУ «Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии им. Кулакова В.И.» Минздрава РФ
Россия
ФГБУ «Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии им. Кулакова В.И.» Минздрава РФ
Россия
Список литературы
1. Wassif CA, Cross JL, Iben J. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med. 2016; 18: 41-48.
2. Михайлова СВ, Захарова ЕЮ, Дегтярева АВ, Печатникова НЛ, Какаулина ВС, Полякова НА. Болезнь Нимана-Пика тип С у детей. Медицинская генетика. 2013; 12 (9): 25-33.
3. Михайлова СВ, Захарова ЕЮ, Петрухин АС. Нейрометаболические заболевания у детей и подростков. Диагностика и подходы к лечению, 2-е издание. 2017.
4. Patterson MC, Mengel E, Wijburg FA. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013; 8: 12.
5. Walterfang M, Bonnot O, Mocellin R. The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis. 2013; 36: 687-702.
6. Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010; 5: 16.
7. Imrie J, Dasgupta S, Besley GT. The natural history of Niemann-Pick disease type C in the UK. J Inherit Metab Dis. 2007; 30: 51-59.
8. Wraith JE, Baumgartner MR. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009; 98 (1-2): 152-165.
9. Patterson MC, Hendriksz ChJ. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Molecular Genetics and Metabolism. 2012; 106 (3): 330-344.
10. Новиков ПВ, Семячкина АН, Воинова ВЮ, Захарова ЕЮ. Федеральные клинические рекомендации по диагностике и лечению болезни Ниманна-Пика тип С. 2013.
11. Pineda М. Suspicion Index to aid screening of early-onset Niemann-Pick disease Type C (NPC), BMC Pediatrics. 2016; 16: 107.
12. Patterson MC, Clayton P, Gissen P. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Practice. 2017; 7 (6): 499-511.
13. Hendriksz CJ. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Current Medical Research and Opinion. 2017; 33(5): 877-890.
14. Strupp M, Kremmyda O, Adamczyk C. Central ocular motor disorders, including gaze palsy and nystagmus. J Neurol. 2014; 261 (2): 542-558.
15. Stone J, Griffiths TD, Rastogi S. Non-Picks frontotemporal dementia imitating schizophrenia in a 22-year-old man. J Neurol 2003; 250: 369-370
16. Synofzik M, Fleszar Z, Schols L. Identifying Niemann-Pick type C in early-onset ataxia: two quick clinical screening tools. J Neurol. 2016; 263: 1911-1918.
17. Schicks J, Muller Vom Hagen J, Bauer P. Niemann-Pick type C is frequent in adult ataxia with cognitive decline and vertical gaze palsy. Neurology. 2013; 80: 1169-1170.
18. Anheim M, Lagha-Boukbiza O, Fleury-Lesaunier MC. Heterogeneity and frequency of movement disorders in juvenile and adult-onset Niemann-Pick C disease. J Neurol. 2014; 261: 174-179.
19. Koens LH, Kuiper A, Coenen MA. Ataxia, dystonia and myoclonus in adult patients with Niemann-Pick type C. Orphanet J Rare Dis. 2016; 11: 121.
20. Sevin M, Lesca G, Baumann N. The adult form of NiemannPick disease type C. Brain. 2007; 130: 120-133.
21. Bauer P, Balding DJ, Klunemann HH. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet. 2013; 22: 4349-4356.
22. Mazzacuva F, Mills P, Mills K. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett. 2016; 590: 1651-1662.
23. Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol. 2015; 15: 257.
24. Nunn K, Williams K, Ouvrier R. The Australian childhood dementia study. Eur Child Adolesc Psychiatry. 2002; 11: 63-70.
25. Saito Y, Suzuki K, Nanba E. Niemann-Pick type C disease: accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity. Ann Neurol. 2002; 52: 351-355.
26. Jiang X, Sidhu R, Mydock-McGrane L. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci Transl Med. 2016; 8: 337-363.
27. Garver WS, Francis GA, Jelinek D. The national NiemannPick C1 disease database: report of clinical features and health problems. Am J Med Genet. 2007; 143 (A): 1204-1211.
28. Stampfer M, Theiss S, Amraoui Y. Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J Rare Dis. 2013; 8: 35.
29. Klarner B, Klunemann HH, Lurding R. Neuropsychological profile of adult patients with Niemann-Pick C1 (NPC1) mutations. J Inherit Metab Dis. 2007; 30: 60-67.
30. Wijburg FA, Sedel F, Pineda M. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology. 2012; 78: 1560-1567.
31. Hendriksz CJ, Pineda M, Fahey M. The Niemann-Pick disease Type C suspicion index: development of a revised tool with improved predictive ability. J Inherit Metab Dis. 2014; 37: 372.
32. Gourzis P, Skokou M, Polychronopoulos P. Frontotemporal dementia, manifested as schizophrenia, with decreased heterochromatin on chromosome 1. Case Rep Psychiatry. 2012; 2012: 1-5.
33. Cooper JJ, Ovsiew F. The relationship between schizophrenia and frontotemporal dementia. J Geriatr Psychiatry Neurol. 2013; 26: 131-137.
34. Klunemann HH, Santosh PJ, Sedel F. Treatable metabolic psycho-ses that go undetected: what Niemann-Pick type C can teach us. Int J Psychiatry Clin Pract. 2012; 16: 162-169.
35. Webber D, Klunemann HH. Psychiatric manifestations of Niemann-Pick disease. J Inherit Metab Dis. 2011; 4: 25-31.
36. Degtyareva AV, Mikhailova SV, Zakharova EY, Tumanova EL, Puchkova AA. Visceral symptoms as a key diagnostic sign for the early infantile form of Niemann-Pick disease type C in a Russian patient: a case report. Journal of Case Reports. 2016; 10: 22-27.
37. Дегтярева АВ, Мухина ЮГ, Дегтярев ДН. Синдром холестаза у новорожденных детей, пособие для врачей. 2011.
38. Hegarty R, Hadzic N, Gissen P. Inherited metabolic disorders presenting as acute liver failure in newborns and young children: King’s College Hospital experience. Eur J Pediatr. 2015; 174: 1387-1392
39. Yerushalmi B, Sokol RJ, Narkewicz MR. Niemann-pick disease type C in neonatal cholestasis at a North American Center. J Pediatr Gastroenterol Nutr. 2002; 35: 44-50.
40. McKay Bounford K, Ruth N, Yeung A. A gene sequencing assay to determine the frequency of genetic conditions associated with cholestasis in neonates and infants. Manuscript in preparation. 2016.
41. Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T, Kolb SA. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol Genet Metab. 2016, 118: 244-254.
42. Каменец ЕА, Милованова НВ, Иткис ЮС, Зубович АИ, Строкова ТВ, Серебреникова ТЕ, Дегтярева АВ, Никитина НВ, Захарова ЕЮ. Применение технологии таргетного секвенирования при диагностике наследственных гепатопатий. Российский вестник перинатологии и педиатрии. 2016; 61 (4): 194.
43. Прошлякова ТЮ, Байдакова ГВ, Букина ТМ, Михайлова СВ, Ильина ЕС, Руденская ГЕ, Клюшников СА, Малахова ВА, Захарова ЕЮ. Биохимические маркеры при болезни Ниманна-Пика тип С. Медицинская генетика. 2015; 8: 3-6.
44. Boenzi S, Deodato F. A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin Chim Acta. 2014; 437: 93-100.
45. Polo G, Burlina AP, Kolamunnage TB, Zampieri M, Dionisi-Vici C, Strisciuglio P, Zaninotto M, Plebani M, Burlina AB. Diagnosis of sphingolipidoses: a new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clinical chemistry and laboratory medicine. 2017; 55: 403-414.
46. Крылова ТД, Прошлякова ТЮ, Байдакова ГВ, Иткис ЮС, Куркина МВ, Захарова ЕЮ. Биомаркеры в диагностике и мониторинге лечения болезней клеточных органелл. Медицинская генетика. 2016; 15 (7): 3-10.
47. Прошлякова ТЮ, Байдакова ГВ, Каменец ЕА, Михайлова СВ, Малахова ВА, Захарова ЕЮ. Оксистеролы в дифференциальной диагностике лизосомных болезней накопления. Медицинская генетика. 2016; 15 (12): 37-41.
Болезнь Ниманна-Пика ( Липоидный гистиоцитоз , Нелейкемический ретикулез , Сфингомиелиновый липидоз , Сфингомиелиноз , Фосфатидоз )
Болезнь Ниманна-Пика – это редкое наследственное заболевание, характеризующееся накоплением липидов в различных органах и тканях, что приводит к нарушению их функций. Отличительной особенностью является выраженный клинический полиморфизм. Наиболее частыми считаются очаговые неврологические симптомы, задержка нервно-психического развития, гепато- и спленомегалия. В диагностике используется определение активности специфических ферментов, гистологические исследования, церебральная томография, молекулярно-генетический анализ. Для лечения применяются симптоматическую, субстрат-редуцирующую терапию.
МКБ-10
Общие сведения
Болезнь Ниманна-Пика (БНП, сфингомиелиноз, сфингомиелиновый липидоз) относится к группе лизосомных болезней накопления. Впервые нозология была описана немецким педиатром А. Ниманном в 1914 г., в 1930 г. немецкий патологоанатом Л. Пик опубликовал патоморфологические данные. Выделяют 3 типа заболевания, различающиеся по патогенезу, эпидемиологии, характеру течения. Распространенность типов А и B среди общей популяции составляет 1 случай на 250 000 человек, типа С – 1 на 120-150 000 населения. У евреев-ашкенази тип А встречается намного чаще, по разным данным 1:40 000-1:100 000.
Причины
В основе возникновения всех разновидностей болезни Ниманна-Пика лежат генетические мутации. Типы А и B вызваны мутацией гена SMPD-I, расположенного в локусе 11p15.4-p15.1. Это ген кодирует энзим кислую сфингомиелиназу. Причиной типа С являются мутации генов NPC1 (локус 18q11-q12) и NPC2 (локус 14q24). Данные гены кодируют белки-переносчики, участвующие в транспорте холестерина и других липидов внутри клетки. Патология наследуется по аутосомно-рецессивному типу.
Патогенез
Механизмы развития заболевания на патогенетическом уровне при разных типах болезни Ниманна-Пика несколько различаются. В результате генетической мутации при БНП-А возникает практически полная недостаточность кислой сфингомиелиназы, что ведет к быстрому накоплению сфинголипидов в ЦНС и других внутренних органах.
Это сопровождается стремительным развитием грубой неврологической симптоматики и летальным исходом уже в раннем детском возрасте. Другая разновидность мутации того же гена при типе B вызывает лишь 20%-е снижение функциональной активности сфингомиелиназы. Поэтому отложение липидов происходит преимущественно в клетках ретикулоэндотелиальной системы (печень, селезенка).
При болезни Ниманна-Пика типа С из-за нарушения работы белков-транспортеров в клетках накапливаются разные классы липидов – неэстерифицированный холестерин, сфингомиелин, гликосфинголипиды. Поражается нервная система, внутренние органы. Агрегации холестерина вызывают вторичное снижение активности сфингомиелиназы за счет подавления ее синтеза.
Классификация
В клинической практике выделяются 3 основные разновидности БНП:
- Тип А (классический инфантильный). Самая тяжелая форма. Характеризуется ранним началом, прогрессирующим течением, быстрым наступлением смерти.
- ТипB(висцеральный). Типично более умеренное течение, поздний дебют. Неврологические симптомы практически отсутствуют.
- Тип С. Наиболее распространенный вид с крайне разнообразной симптоматикой. В зависимости от возраста начала манифестации подразделяется на следующие формы:
- неонатальная – до 3 месяцев;
- ранняя младенческая – от 3 месяцев до 2 лет;
- поздняя младенческая – от 2 до 6 лет;
- юношеская (ювенильная) – от 6 до 15 лет;
- взрослая – старше 15 лет.
Симптомы болезни Нимана-Пика
Тип А
Первые признаки появляются почти с самого рождения – это увеличение печени, селезенки, лимфоузлов. С 4-6 месяцев у ребенка снижается аппетит, присоединяются тошнота, рвота. Такие базовые навыки, как способность удерживать голову, ходьба, речь, значительно задерживаются. На втором году жизни формируется спастичность мышц. При глубоком нейродегенеративном поражении головного мозга развиваются нарушение дыхания и сердцебиения, что является основной причиной смерти.
Тип В
Для данной разновидности неврологические симптомы не характерны. Основные симптомы – гепатоспленомегалия, генерализованная лимфаденопатия, частые респираторные инфекции. Поражение дыхательной системы несет наибольшую угрозу для жизни. Инфильтрация альвеол приводит к формированию интерстициальной патологии легких к 20-25 годам, поэтому пациенты испытывают серьезные проблемы с дыханием.
Тип С
Данный тип болезни Ниманна-Пика отличается широким спектром клинических симптомов. Наиболее часто заболевание дебютирует с 7-12 лет. Помимо задержки общего развития имеется снижение общего мышечного тонуса, нарушение походки, координации движений, часто возникают падения. Специфичным неврологическим признаком является ограничение движения глаз при взгляде вверх и вниз.
Еще одним патогномоничным, но редким симптомом считается геластическая катаплексия – внезапная потеря мышечного тонуса в ногах, руках или шее, которая провоцируется эмоциями, например смехом. Ребенок испытывает затруднения при произношении слов или звуков, речь становится невнятной, неразборчивой. Возможны непроизвольные болезненные спазмы мышц лица или кистей рук. Вследствие нарушенного глотания часто возникают поперхивания при приеме пищи.
Нередко происходят тонико-клонические, генерализованные эпилептические припадки. У ребенка значительно ухудшается способность к обучению, запоминанию, быстро утрачиваются недавно приобретенные навыки. У 25% пациентов встречаются острые психозы с галлюцинациями. Иногда отмечаются депрессия, биполярное, обсессивно-компульсивное расстройство (ОКР). Из симптомов поражения внутренних органов характерна гепатоспленомегалия с холестазом.
Осложнения
Для заболевания характерно большое количество осложнений. К наиболее опасным относятся остановка дыхательной или сердечной деятельности вследствие поражения глубинных структур головного мозга. При неонатальной форме БНП типа С быстро прогрессирует печеночная, дыхательная недостаточность, высока вероятность развития водянки плода.
Из-за снижения тонуса мышц глотки возможно попадание пищи в дыхательные пути (аспирация). Инфильтраты в легких способствуют возникновению пневмоний, увеличению давления в сосудах малого круга кровообращения, формированию легочного сердца (правожелудочковой сердечной недостаточности). Отложение липидов в печеночной ткани может привести к циррозу печени.
Диагностика
Курацией больных с БНП занимаются врачи-педиатры, неврологи. При физикальном обследовании важен неврологический осмотр – оценка мышечного тонуса, сухожильных рефлексов, мозжечковых проб. Дифференцировать болезнь Ниманна-Пика необходимо с болезнью Гоше, Тея-Сакса, Вильсона-Коновалова. Для уточнения диагноза назначаются дополнительные методы исследования:
- Рутинные лабораторные исследования. В АОК часто отмечается снижение количества тромбоцитов, реже – гемоглобина, эритроцитов, лейкоцитов. В биохимическом анализе крови обнаруживается увеличение концентрации печеночных трансаминаз (АЛТ, АСТ), билирубина, холестерина.
- Специфические лабораторные тесты. При типах А, B БНП в лейкоцитах выявляется уменьшение уровня кислой сфингомиелиназы. При типе С в крови резко повышены активность хитотриозидазы, содержание продуктов окисления холестерина – 7-кетостерола, холестан-3,5,6-триола.
- Томографические методы. На КТ или МРТ головного мозга визуализируется атрофия коры больших полушарий и мозжечка, истончение мозолистого тела, умеренное расширение желудочков.
- Гистологические исследования. При окраске биоптата кожи филипином наблюдаются интенсивно светящиеся области, сконцентрированные вокруг ядер, которые представляют собой скопления неэстерифицированного холестерина. В аспирате костного мозга отмечается инфильтрация пенистыми клетками (клетками Ниманна-Пика), лазурные гистиоциты.
- ДНК-диагностика. Самый точный метод исследования, позволяющий достоверно подтвердить болезнь Ниманна-Пика – молекулярно-генетическое тестирование, при котором определяются мутации генов SMPD-1, NPC-1, NPC-2.
Лечение болезни Ниманна-Пика
Консервативная терапия
Всем пациентам показана обязательная госпитализация в стационар. Специфическая терапия БНП-А и БНП-В пока не разработана, проводится только симптоматическое лечение. Для прерывания начального звена патогенеза БНП-С назначается субстрат-редуцирующая терапия – препарат миглустат, блокирующий начальные этапы синтеза гликосфинголипидов.
Таким образом значительно уменьшается накопление сфинголипидов в тканях. Благодаря приему миглустата удается добиться замедления прогрессирования и регресса неврологической симптоматики. Препараты для симптоматической терапии всех видов болезни Ниманна-Пика следующие:
- Противосудорожные. Для предупреждения эпилептических припадков назначаются антиконвульсанты – карбамазепин, вальпроевая кислота, ламотриджин.
- Психотропные. С целью коррекции психических расстройств применяются нейролептики (хлорпротиксен), селективные ингибиторы обратного захвата серотонина (флуоксетин).
- Холиноблокаторы и миорелаксанты. Пациентам с дистонией и мышечными спазмами целесообразно использование биперидена, баклофена, тизанидина.
- Желчегонные. Для борьбы с внутрипеченочным холестазом эффективным препаратом является урсодезоксихолевая кислота.
- Антидиарейные ЛС и спазмолитики. При развитии диспепсических симптомов на фоне приема миглустата дополнительно назначается лоперамид, дротаверин.
- Статины. Для снижения уровня холестерина в крови применяется аторвастатин или розувастатин.
Хирургическое лечение
Оперативные вмешательства оказались успешными только в случае болезни Ниманна-Пика-В. Трансплантация стволовых клеток костного мозга у части пациентов позволяет уменьшить степень висцеральных симптомов – гепатоспленомегалии, интерстициального поражения легких. При гиперспленизме с панцитопенией выполняется спленэктомия. Развитие цирроза печени с тяжелой печеночной недостаточностью служит показанием к пересадке печени.
Экспериментальное лечение
Продолжаются исследования по поиску эффективных методов лечения болезни Ниманна-Пика. При экспериментах на лабораторных мышах под влиянием генной терапии в клетках повышалась активность сфингомиелиназы. В настоящее время на стадии клинических испытаний находится препарат 2-гидроксипропил-бетациклодекстрин и заместительная ферментотерапия БНП типа В.
Паллиативное лечение
На позднем этапе заболевания с запущенным нейродегенеративным процессом применяются меры по облегчению состояния больного. При выраженном нарушении глотания может возникнуть необходимость в зондовом питании или наложении гастростомы для того, чтобы обеспечить пациента достаточным количеством питательных веществ и жидкости.
Прогноз и профилактика
В большинстве случаев прогноз для жизни при болезни Ниманна-Пика неблагоприятный. Относительно доброкачественным считается тип В, при котором не затрагивается нервная система. При типе А продолжительность жизни составляет 1-4 года, при типе C – около 10-20 лет с момента постановки диагноза. Наиболее частыми причинами смерти выступают поражения мозговых структур, регулирующих дыхательную и сердечную деятельность.
Реже летальный исход наступает от тяжелых инфекций дыхательных путей, печеночной недостаточности. Основным методом первичной профилактики является пренатальная диагностика на ранних сроках беременности. В ворсинах хориона молекулярно-генетическими тестами определяется наличие мутаций NPC1, NPC2, SMPD-1; в амниоцитах исследуется активность сфингомиелиназы.
1. Болезнь Ниманна-Пика, тип С - лизосомная патология с нарушением внутриклеточного транспорта липидов/ Клюшников С.А.// Нервные болезни. - № 1. - 2014.
2. Болезнь Ниманна-Пика типа С. Клинические примеры/ Михайлова С.В., Захарова Е.Ю., Букин Т.М., Савина Д.А., Пилия С.В., Петрухин А.С.// Педиатрическая фармакология. - Том 7, №5 - 2010.
3. Болезнь Ниманна-Пика тип С: молекулярные механизмы патогенеза и подходы к лечению/ Михайлова С.В., Захарова Е.Ю., Губина Е.Б., Савин Д.А., Ильина Е.С., Пилия С.В., Печатникова Н.Л., Бологов А.А.// Эффективная фармакотерапия. Педиатрия, спецвыпуск «Неврология». - 2011.
Трудный диагноз. Болезнь Ниманна – Пика, тип С

Л.С. НАМАЗОВА-БАРАНОВА 1, 2, 3, А.К. ГЕВОРКЯН 1, 2,, Н.Д. ВАШАКМАДЗЕ 1, Л.С. ВЫСОЦКАЯ 1, А.М. МАМЕДЬЯРОВ 1, д.м.н., Т.В. МАРГИЕВА 1, 2
1 Научный центр здоровья детей РАМН, Москва
2 Первый Московский государственный медицинский университет им. И.М. Сеченова
3 Российский научный исследовательский медицинский университет им. Н.И. Пирогова, Москва
В условиях современных диагностических возможностей и повышения уровня знаний врачей все чаще выявляют болезни, которые ранее считались крайне редкими. Наряду с достижениями фармакологической промышленности, своевременная диагностика и назначение адекватной терапии позволяют во многих случаях сохранить жизнь ребенку и замедлить прогрессирование болезни. Статья посвящена редкой, генетически обусловленной патологии из группы лизосомных болезней, наследуемой аутосомно-рецессивно, — болезни Ниманна – Пика, тип С. Подробно представлены варианты клинического течения и методы диагностики.
Болезни накопления — группа редких генетически детерминированных болезней, обусловленных нарушением синтеза лизосомных ферментов, контролирующих внутриклеточное расщепление макромолекул (гликозаминогликанов, гликолипидов, гликопротеинов) и внутрилизосомным их накоплением, что приводит к прогрессирующему нарушению функции поврежденных органов. Наиболее часто в патологический процесс вовлекаются печень, селезенка, центральная нервная система, костная система [1–4]. В результате мутации гена снижается активность того или иного фермента (до 10–20% нормы), что характерно для мукополисахаридозов и гликогенозов. В редких случаях нарушается т. н. сортировка молекул внутри клеток. Последнее свойственно болезни Ниманна – Пика, тип С (НП-С), которая представляет собой сфингомиелиновый липидоз, развивающийся в результате наследственной недостаточности белков, участвующих во внутриклеточном транспорте липидов, и вторичного снижения активности сфингомиелиназы (до 20% нормы). Заболевание наследуется по аутосомно-рецессивному типу. Гены NPC1 и NPC2 локализуются в локусах 18q11-q12 и 14q24.3. Болезнь обусловлена мутацией гена NPC1 (95%) или NPC2 (4%), в 1% случаев молекулярно-генетический дефект идентифицировать не удается [1–9].
Классификация
В зависимости от молекулярно-генетического дефекта выделяют два типа заболевания: болезнь Ниманна – Пика, тип С, 1-го типа и болезнь Ниманна – Пика, тип С, 2-го типа. Первоначально выделяли в отдельную форму болезнь Ниманна – Пика, тип D, выявляемую в изолятах Новой Шотландии, но в дальнейшем было показано, что причиной этой формы болезни также являются мутации гена NPC1 [2, 5, 10].
Эпидемиология
НП-С относится к числу редких наследственных болезней обмена веществ. Заболевание распространено повсеместно. Частота заболевания — 1 : 120 000–1 : 150 000 живых новорожденных. Высокая частота болезни НП-С 1-го типа отмечена среди некоторых генетических изолятов: французской колонии Акадия (Новая Шотландия), групп бедуинов в Израиле, испанских поселений в Колорадо и Нью-Мексико (США), что связано с эффектом основателя [11, 12].
Клинические формы болезни Ниманна – Пика, тип С
Болезнь может манифестировать в любом периоде жизни — от внутриутробного до пожилого возраста, но чаще признаки НП-С появляются в раннем детском возрасте и быстро прогрессируют, приводя к нарушению функций поврежденных органов. Течение болезни и продолжительность жизни пациента во многом зависят от возраста, в котором произошел дебют. Выделяют следующие формы НП-С: неонатальную, младенческую, позднюю младенческую, юношескую и взрослую [2, 11–13].
Неонатальная форма (дебют до 3 мес.) характеризуется развитием внутриутробной водянки плода. После рождения преобладает мышечная гипотония и задержка психомоторного развития, которые прогрессируют и приводят к нарушениям интеллекта. Приблизительно в половине случаев отмечаются внутрипеченочный холестаз, желтуха и гепатоспленомегалия, которые с возрастом могут полностью исчезнуть. Редко наблюдается молниеносное течение с развитием тяжелой формы холестатической желтухи и неблагоприятным исходом на первом полугодии жизни от печеночной недостаточности. В небольшом числе случаев развивается дыхательная недостаточность [9, 10, 13].
При младенческой форме (3 мес. – 2 года) основные клинические симптомы НП-С (гепатоспленомегалия, задержка психомоторного развития, мышечная гипотония) появляются на первом году жизни. Большинство пациентов никогда не приобретают навыки самостоятельной ходьбы. Интеллектуальное развитие обычно не страдает. По мере прогрессирования болезни происходит вовлечение в патологический процесс пирамидной системы, постепенно развивается снижение когнитивных функций [11].
НП-С в 60–70% случаев обычно манифестирует в позднем младенческом и раннем детском возрасте (поздняя младенческая форма — 2–6 лет). Первыми клиническими симптомами этой формы являются мозжечковые расстройства (шаткость при ходьбе, дизартрия, дисметрия), которые обычно проявляются в возрасте 3–5 лет. В 90% случаев наблюдается гепатоспленомегалия различной степени выраженности. По мере прогрессирования болезни (в т. ч. при юношеской форме) практически у всех пациентов развивается вертикальный супрануклеарный офтальмопарез [14]. На начальных этапах происходит замедление движения глазных яблок по вертикали, постепенно прогрессирующее до полного ограничения вертикального, а иногда и горизонтального взора. Нередко отмечается задержка речевого развития. В дальнейшем постепенно утрачиваются ранее приобретенные двигательные навыки, проявляются интеллектуальные нарушения, развивается дисфагия и дизартрия, реже демиелинизирующая периферическая полинейропатия [14, 15]. Неблагоприятный исход при поздней младенческой форме обычно наступает в первую декаду жизни [2, 13].
При юношеской форме болезни НП-С первые симптомы обычно появляются в возрасте от 6 до 15 лет в виде нарушения усвоения школьного материала, письма, снижения внимания, памяти, гиперактивного поведения, что приводит к установлению неправильного диагноза. В некоторых случаях болезнь манифестирует с психиатрических нарушений, таких как нарушение поведения, шизофреноподобный синдром, депрессия [2, 11]. В 20% случаев наблюдается геластическая катаплексия: кратковременная приступообразная утрата мышечного тонуса, приводящая к падению больного без потери сознания, чаще возникающая на фоне сильных эмоциональных реакций (например, во время смеха) [16, 17]. Со временем прогрессируют мозжечковые расстройства, появляются дисфагия, дизартрия и ухудшается интеллектуальное развитие. Частыми симптомами НП-С являются экстрапирамидные расстройства в виде различных дистонических гиперкинезов. В половине случаев развиваются фокальные и/или генерализованные эпилептические приступы, трудно поддающиеся адекватной антиэпилептической терапии. На поздних стадиях НП-С нарастают пирамидные нарушения в виде повышения мышечного тонуса, оживления сухожильных рефлексов, появления патологических рефлексов, бульбарно-псевдобульбарного синдрома; развивается деменция, децеребрационная или декортикационная ригидность. Гепатоспленомегалия обычно не наблюдается. Неблагоприятный исход чаще развивается в конце второго – начале третьего десятилетия жизни, обычно от интеркуррентных инфекций [16–18].
Взрослая форма. Симптомы болезни НП-С появляются на втором-третьем десятилетии жизни, но могут возникнуть и в возрасте старше 50 лет [18]. Характерно медленно прогрессирующее течение болезни. Постепенно развиваются мозжечковые расстройства (атаксия, дизартрия, дисметрия), мышечная дистония, различной степени выраженности интеллектуальные расстройства. Нередко у взрослых болезнь НП-С манифестирует с развития биполярных расстройств [16–18]. При этой форме эпилептические приступы развиваются редко, не характерно увеличение селезенки и вертикальный надъядерный офтальмопарез.
Диагностика
В настоящее время основными методами диагностики НП-С считают биохимические тесты, позволяющие выявить нарушение внутриклеточного транспорта и гомеостаза холестерина. Наличие мутаций NPC1- или NPC2- генов в сочетании с характерными клиническими проявлениями подтверждает диагноз. Для изучения новых мутаций необходимы дополнительные исследования [1, 2, 5, 6, 9].
Для диагностики болезни биохимическими методами требуется наличие живых клеток (обычно культуры фибробластов кожи).
Самым чувствительным и специфичным методом считают тест с окрашиванием филипином. Фибробласты культивируют в среде с большим содержанием липопротеидов низкой плотности, после чего клетки фиксируют и окрашивают филипином. В 85% случаев («классический биохимический фенотип») при флюоресцентной микроскопии в исследуемых клетках обнаруживают многочисленные флюоресцирующие (заполненные холестерином) перинуклеарные пузырьки. Менее выраженное накопление холестерина отмечается в 15% случаев («вариантный биохимический фенотип»). Интерпретировать результаты микроскопии в таких случаях часто трудно, а результаты могут быть как ложноположительными, так и ложноотрицательными [19, 20].
Второй по информативности тест — измерение скорости образования эфира холестерина, индуцированного липопротеинами низкой плотности, в культуре фибробластов [19, 20]. В клеточных линиях пациентов с «классическим биохимическим фенотипом» скорость этерификации холестерина близка или равна нулю; при «вариантном биохимическом фенотипе» наблюдается легкое нарушение этерификации холестерина. Соответственно, результаты теста могут оказаться неоднозначными. В таких случаях для подтверждения диагноза необходим анализ генетических мутаций. Учитывая сложность, высокую стоимость и трудоемкость биохимического исследования, при положительной пробе с филипином часто сразу проводят анализ мутаций [19, 20].
Биохимические тесты недостаточно информативны в случае гетерозиготных носителей болезни НП-С: результаты пробы с филипином при этом могут быть нормальными; наблюдаются легкие нарушения, сходные с таковыми в «вариантных» клеточных линиях [20, 21].
Исследование мазков костного мозга, окрашенных филипином (а также по Гимзе), позволяет выявить пенистые клетки, заполненные холестерином, считается экспресс-методом скрининга НП-С, однако не дает возможности установить окончательный диагноз [20, 21].
Для пренатальной диагностики НП-С следует использовать молекулярно-генетический метод [5].
Гистологические методы предполагают исследование биоптатов тканей с помощью световой и электронной микроскопии. Обнаруживают характерные (но неспецифические) пенистые клетки и/или голубые гистиоциты в костном мозге, селезенке, печени, легких и лимфатических узлах (при световой микроскопии), в некоторых случаях выявляя скрытое поражение кожи, скелетных мышц и глаз [16, 22, 23]. Отсутствие изменений при световой микроскопии не исключает диагноз болезни НП-С (например, проявляющейся холестатическим поражением печени у новорожденных).
Патогномонично для НП-С обнаружение полиморфных цитоплазматических включений при электронной микроскопии биоптатов печени или кожи, однако, их выявляют не всегда [5, 16, 22].
Генетическое исследование является золотым стандартом для окончательной верификации диагноза, а также для пренатальной диагностики [5, 16]. Молекулярно-генетическое исследование генов NPC1 и NPC2 проводится в специализированных лабораториях. В некоторых случаях для выявления мутации NPC1 необходимо выполнить комбинированное исследование gДНК и cДНК [5, 6, 16, 23].
Выявление носителей и пренатальный диагноз
В случае подтверждения диагноза болезни НП-С необходимо осуществить генетическое консультирование пациента: уточнить характер заболевания, тип наследования, дать рекомендации по планированию семьи (с проведением пренатальной диагностики). ДНК рекомендовано исследовать у обоих родителей.
Пренатальный диагноз может быть установлен на основании исследования хорионических ворсинок на 10–12-й нед беременности. Предпочтительным считают молекулярно-генетический анализ, для которого (в отличие от биохимической пренатальной диагностики) не требуется культура клеток [5].
Поскольку время начала специфической и адекватной симптоматической терапии имеет решающее значение для прогноза при болезни НП-С, важно как можно раньше выявить болезнь — установить диагноз. Данная проблема побудила группу исследователей во главе с профессором F.A. Wijburg к созданию алгоритма диагностики болезни НП-С [29].
Ученые поставили перед собой следующие цели:
1) разработать прогностический инструмент «индекс подозрения болезни НП-С» и валидировать специфичность и чувствительность показателей на основании анализа историй болезней пациентов;
2) разработать балльную шкалу оценки симптомов и их комбинаций для оценки вероятности диагноза болезни НП-С;
3) разработать простой инструмент, который мог бы позволить врачам различных специальностей, не знакомым с проблемой болезни НП-С:
— понимать основные симптомы и проявления,
— предположить у пациента болезнь НП-С как возможный диагноз.
В рамках данного исследования все симптомы болезни НП-С были разделены на три основные категории: висцеральные, неврологические и психиатрические. Ретроспективный анализ данных 216 пациентов, направленных на обследование с подозрением на болезнь НП-С в 7 центров Европы и Австралии, позволил оценить диагностическую важность каждого отдельного симптома и их совокупности. По результатам статистического анализа, симптомы внутри каждой категории были разделены на 5 групп по степени вероятности болезни НП-С: от наиболее вероятных (40 баллов/пункт), таких как вертикальный супрануклеарный паралич взора и геластическая катаплексия, до маловероятных или дополнительных (1 балл/пункт), таких как гипотония, судороги и миоклонус. К примеру, атаксия встречается примерно у 80% пациентов с болезнью Ниманна – Пика, тип С [1], как симптом в данном алгоритме оценена в 10 баллов (средний индекс вероятности). Подобный симптом может встречаться при многих других наследственных болезнях, то есть специфичность его невысока. В то же время необъяснимая изолированная спленомегалия, встречающаяся примерно в 50% случаев болезни НП-С [20], является высокоспецифичным симптомом этой патологии (20 баллов).
В разделе «висцеральные симптомы» учитываются также данные анамнеза. Несмотря на возможность полного разрешения в течение первого года жизни затяжной желтухи, гепатоспленомегалии (в случае их наличия у пациентов в периоде новорожденности), эти симптомы являются важными для постановки диагноза.
В ходе разработки диагностического алгоритма была проведена оценка взаимосвязи симптомов. Наличие у пациента одновременно симптомов из категорий «неврологические» и «висцеральные» повышает прогностический балл НП-С на 40, как и сочетание висцеральных и психиатрических симптомов. Сочетание неврологических и психиатрических симптомов оценено в 20 баллов. Дополнительно оценивается семейный риск: 40 баллов — при наличии родных братьев/сестер или родителей с болезнью Ниманна – Пика, тип С, 20 баллов — двоюродных братьев/сестер с данной патологией.
Суммарная оценка или прогностический балл позволяет врачу разработать тактику дальнейшего ведения пациента:
Читайте также: