Синдром фон Гиппеля-Линдау на МРТ
Обновлено: 27.04.2024
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Болезнь фон Гиппеля-Линдау (VHL-синдром)
ФГУ Эндокринологический научный центр, Москва
Болезнь фон Гиппеля-Линдау является наследственным опухолевым синдромом, предполагающим развитие различных доброкачественных и злокачественных новообразований (гемангиобластома центральной нервной системы и сетчатки глаза, опухоль внутреннего уха, карцинома и кисты почек, феохромоцитома, нейроэндокринная опухоль и кисты поджелудочной железы, цистаденома придатка яичка у мужчин и широкой связки у женщин). Болезнь фон Гиппеля-Линдау - наиболее распространенная причина наследственного рака почки.
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
В 1895 г. Ю. фон Гиппель [1] описал пациента с ретинальной ангиомой, а в 1926 г. А. Линдау [2] — пациента с ретинальной ангиомой и гемангиоматозом центральной нервной системы. Год спустя тот же автор обнаружил ассоциацию этих проявлений с почечными и панкреатическими кистами [3]. Термин «синдром von Hippel—Lindau» (VHL) был введен Мелмоном и Роузеном [4]. Этот синдром выявляется приблизительно у 1 из 36 000 человек [5] и обусловлен мутацией в участке 3p25/26, где локализован ген подавления роста опухоли VHL [6—8]. 23% пациентов не имеют семейного анамнеза заболевания [9—13].

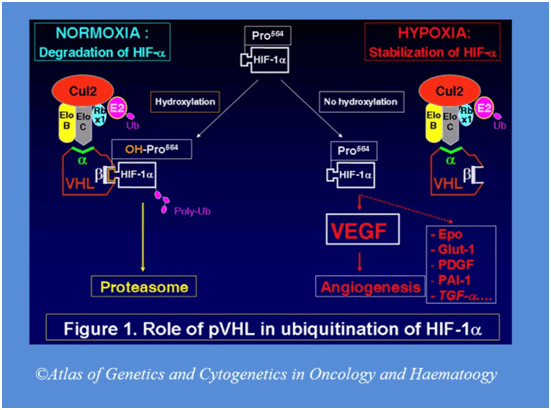
Ген VHL был идентифицирован в 1993 г. [8, 14]. Приблизительно у 20% пациентов выявляется делеция VHL-локуса в материнской или отцовской аллели [15, 16]. Герминальные мутации VHL наследуются по аутосомно-доминантному типу. Почти все мутации VHL у пациентов с феохромоцитомой являются миссенс-мутациями. Ген VHL состоит из 3 экзонов. Белок VHL (pVHL) включает 213 аминокислотных остатков, и его молекулярная масса равна приблизительно 28 кДa. Клетки с дефицитом pVHL накапливают фактор, индуцирующий гипоксию (HIF), что приводит к перепроизводству HIF-зависимых продуктов (которые вовлечены в адаптацию к гипоксии): сосудистого эндотелиального фактора роста (VEGF), эритропоэтина и трансформирующего ростового фактора альфа (TGF). Это объясняет сильную васкуляризацию VHL-ассоциированных опухолей [17—19]. Таким образом, продукт мутированного гена VHL приводит к сверхрегулированию различных генов, участвующих в патогенезе гипоксии, ускоряет ангиогенез, изменяет внеклеточный матрикс и регуляцию клеточного цикла [20—28]. Однако точные механизмы туморогенеза при синдроме VHL в настоящее время остаются неизвестными (рис. 1). Рисунок 1. Комплекс VBC (VHL protein and Elongin B, C) и механизм его действия. а — Normoxic condition — нормальное давление кислорода; Ub — убиквитин; HIF — фактор, индуцирующий гипоксию; pVHL — VHL-протеин; α — элонгин C-связывающий домен pVHL; β — β-домен (субстратсвязывающий домен) pVHL; CUL2 — куллин-2, формирующий комплекс с элонгином-B, C и pVHL; б — мутация гена VHL и, как результат, отсутствие регуляции HIF, VEGF, PDGF и TGF-α; в — мутация гена VHL и отсутствие регуляции aPKC λ (атипичная протеинкиназа C) [29].
VHL-синдром характеризуется развитием гемангиобластом сетчатки глаза (ангиомы сетчатки) и центральной нервной системы (ЦНС), билатеральной и мультифокальной дифференцированной карциномы почки, поликистоза почек, феохромоцитомы, кист и нейроэндокринных опухолей поджелудочной железы, папиллярной цистаденомы придатка яичка у мужчин и широкой связки у женщин, опухолей внутреннего уха. Поражение различных органов и степень этого поражения очень вариабельны (табл. 1, 2).

Клинически заболевание делится на две группы. Тип 1 включает главным образом большие делеции или мутации гена VHL и характеризуется полным фенотипом заболевания [поражение сетчатки, кисты или опухоли головного и спинного мозга, панкреатические, почечные, и селезеночные кисты, солидные панкреатические опухоли (реже аденокарциномы), карциномы почек, цистаденомы эпидидимуса и опухоль внутреннего уха], но без феохромоцитомы. Тип 2, при котором развивается феохромоцитома (миссенс-мутации гена VHL может иметь и неполный фенотип [13, 37, 40]. Тип 2 подразделяется на подтипы с низким (тип 2A) и высоким (тип 2B) риском развития рака почки, а также тип 2C, проявляющийся только феохромоцитомой [35, 38, 41—43] (табл. 3).

Гемангиобластомы ЦНС могут выявляться в детском возрасте, однако средний возраст диагностирования составляет 29 лет [32, 45, 46]. VHL-ассоциированные гемангиобластомы выявляются в среднем на 15 лет раньше, чем спорадические [47]. В зависимости от размера и местоположения опухоли клинические признаки гемангиобластомы ЦНС включают головную боль, тошноту, головокружение, атаксию, расстройство координации движений, нистагм, расстройства речи. Гемангиобластома спинного мозга может приводить к слабости конечностей и парестезиям. Диагноз устанавливается с помощью МРТ головного мозга и позвоночника. Гемангиобластомы обычно характеризуются медленным ростом и имеют высокий риск кровотечений, часто являются мультифокальными. Понимание патогенеза заболевания важно для выбора оптимального времени скрининга на опухоли и лечение [19]. Исследование тканей ЦНС умерших пациентов помогло пониманию гистогенеза гемангиобластом [49]. Активация фактора, индуцирующего гипоксию 2-альфа (HIF 2-α) происходит в маленьких мезенхимальных опухолях и в мезенхимальном компоненте больших опухолей. Активация HIF 1-α наблюдается в эпителиальном компоненте. Это позволило предполагать, что поражение ЦНС при VHL-синдроме — длительный процесс гемангиобластической пролиферации и дифференцировки [50] (рис. 2). Рисунок 2. Гемангиобластома ЦНС.

Поражения глаз выявляются примерно у 37% пациентов с VHL-синдромом, среди них только у 14% обнаруживается полная делеция VHL [51, 52]. Приблизительно у 8% пациентов снижена острота зрения [53]. Для лечения ангиомы сетчатки используют лазерную или криотерапию [32, 34, 54]. Недавние исследования [ 55, 56] показали, что при внутривенном введении антагониста сосудистого эндотелиального фактора роста (anti-VEGF) в течение 7 мес размер гемангиобластом не уменьшается (рис. 3). Рисунок 3. Ангиоматоз сетчатки.
У пациентов с синдромом VHL могут встречаться как кисты, так и рак почек [57—62]. Средний возраст манифестации — 37 лет. Для диагностики используют КТ и УЗИ [36, 63, 64]. Поскольку со`лидные раки могут содержать кистозные части (что затрудняет дифференцирование доброкачественных и злокачественных процессов с помощью визуализирующих методик), при отсутствии данных о метастазах лечение должно быть направлено на удаление этих образований по возможности с соблюдением принципа органосохраняющей операции. Это позволяет поддерживать почечную функцию максимально долго и избежать диализа [58, 65]. Опухоли почек отличаются медленным ростом (3 см (по стандартам США) или 5 см (по стандартам Европы) [58, 60, 67, 68]. Некоторые авторы [69] сообщают о высоком риске местного рецидива (приблизительно 50%) в среднем в течение 53 мес (диапазон 10—115 мес) и росте опухоли со скоростью 34 мм/год (диапазон 1—10,8 мм). «Золотым» стандартом лечения небольших опухолей является открытая и лапароскопическая частичная нефрэктомия. В настоящее время используются также альтернативные методы — криотерапия и радиочастотная аблация [70]. Последние методы могут повлиять на результат патоморфологического диагноза, хотя, по некоторым данным, патоморфологический диагноз после первого цикла криотерапии приблизительно в 91% случаев подтверждает результаты предварительной биопсии [71].

Феохромоцитома выявляется примерно у 26% пациентов с синдромом VHL [37]. У пациентов с очевидно спорадической феохромоцитомой в 3—11% случаев впоследствии выявляют мутацию VHL [10, 12, 13]. Феохромоцитома может быть первым проявлением синдрома [30, 72]. В большинстве случаев надпочечниковые феохромоцитомы при VHL-синдроме двусторонние (синхронные или метахронные) [37, 73]. Вненадпочечниковые феохромоцитомы встречаются примерно в 30% случаев [37, 74—76]. Феохромоцитомы как часть синдрома VHL имеют исключительно норадреналиновый фенотип. Биохимические маркеры опухоли могут помочь отличить VHL-ассоциированные феохромоцитомы от феохромоцитом при синдроме МЭН 2-го типа [75]. Различия в биохимическом фенотипе при VHL-синдроме и МЭН 2-го типа связаны с различной экспрессией тирозингидроксилазы (TH) — лимитирующего фермента синтеза катехоламинов, и фенилэтаноламин-N-метилтрансферазы (PNMT). У пациентов с синдромом VHL отмечена низкая экспрессия PNMT, преобразующей норадреналин в адреналин. Различия биохимического фенотипа также связаны с различиями хранения, транспорта и секреции катехоламинов [77]. МЭН 2-ассоциированные феохромоцитомы содержат более высокие концентрации катехоламинов из-за более выраженной экспрессии TH. VHL-ассоциированные феохромоцитомы, секретируют катехоламины непрерывно, тогда как при синдроме МЭН 2 отмечен эпизодический характер секреции. Это определяет и различия клинических проявлений двух синдромов. Например, пациенты с МЭН 2 чаще жалуются на кризовые подъемы АД [78]. Помимо генетических различий [26], регистрируется разная экспрессия эритропоэтина и его рецептора [79]. Кроме того, около 80% феохромоцитом бессимптомны и выявляются случайно при визуализирующих исследованиях. Низкая чувствительность некоторых радионуклидных методов визуализации может объясняться относительной нехваткой гранул хранения или уменьшенной экспрессией мембранного норадреналина или везикулярных моноаминных транспортеров [80]. Поэтому сцинтиграфия с 123 I-MIBG (метайодбензилгуанидином) часто не обнаруживает VHL-связанные надпочечниковые феохромоцитомы [81, 82]. ПЭТ с 6-18F-фтордопамином более чувствительный метод [36, 83]. Злокачественные феохромоцитомы с метастазами в легких, печени, костях, лимфоузлах редко встречаются при синдроме VHL [37, 74, 84—87]. Метастазы выявляются менее чем в 7% случаев [37]. К сожалению, в настоящее время нет четких признаков, позволяющих надежно отличить доброкачественную от злокачественной феохромоцитомы, хотя уже известно, что герминальная мутация гена SDHB, является в этом отношении точным маркером [86—88]. Выявление феохромоцитомы у пациентов с синдромом VHL особенно важно, учитывая высокую вероятность хирургических вмешательств по поводу других опухолей (гемангиобластом ЦНС и др.). Невыявленная феохромоцитома при других вмешательствах может привести к опасным для жизни гипертоническим кризам. Более 70% феохромоцитом у детей являются VHL-ассоциированными. Каждому пациенту с VHL-синдромом и подтвержденной феохромоцитомой до оперативного лечения необходимо проводить ПЭТ с 6-18F-фтордопамином или сцинтиграфию с 123 I-MIBG для выявления вненадпочечниковой феохромоцитомы или метастазов [89]. Лечение феохромоцитомы оперативное. В то же время 6-месячная терапия ингибиторами тирозинкиназы приводит к уменьшению опухоли на 21% и сокращению уровня норметанефринов и хромогранина А в плазме [90] (рис. 4). Рисунок 4. Двусторонняя феохромоцитома и поликистоз почек.
Цистаденомы эпидидимуса — доброкачественные опухоли, которые могут быть двусторонними [48]. Чаще они имеют около 2 см в диаметре, могут распространяться на семенной канатик, приводя к бесплодию. Хирургическое лечение обычно не проводят; необходимо наблюдение [92].
У 35—75% пациентов с синдромом VHL имеются доброкачественные кисты и микрокистозные аденомы поджелудочной железы [93—96]. По данным КТ, у 17% пациентов выявляются панкреатические нейроэндокринные опухоли [95, 97, 98]. Из 633 пациентов с VHL-синдромом у 108 (17%) обнаруживались панкреатические эндокринные опухоли, и у 9 (8%) были выявлены метастазы [99]. Метастазирование более вероятно, если размеры опухоли превышают 3 см. У 78% пациентов с метастазами (7 из 9) мутация локализовалась в экзоне 3 гена VHL, и удвоение массы опухоли в среднем происходило за 337 дней. Таким пациентам необходимо оперативное лечение. Если размеры опухоли меньше 3 см, она растет медленно, а мутация локализована не в экзоне 3, то можно ограничиться наблюдением за пациентом. Средний возраст диагностики нейроэндокринных опухолей поджелудочной железы — 35 лет. Панкреатические кисты встречаются у больных начиная с 15 лет и чаще являются бессимптомными. В зависимости от размера и местоположения клинические симптомы могут быть вызваны обструкцией желчных путей и/или ферментной недостаточностью. В таких случаях устанавливают желчный стент и/или назначают ферментные препараты.
Гормонально-активные нейроэндокринные опухоли встречаются редко [95, 100]. По данным последних исследований, смертность при панкреатических эндокринных опухолях составляет 6%. В 60% случаев при сцинтиграфии обнаруживают рецепторы соматостатина; злокачественные опухоли выявлялись в 58% случаев [101].
Опухоли внутреннего уха располагаются в лабиринте, под твердой мозговой оболочкой на задней поверхности пирамиды височной кости [102—104]. Эти опухоли практически не метастазируют [105]. Симптомы включают потерю слуха, звон в ушах, головокружение и/или парез лицевого нерва [106, 107]. Таким образом, пациентам с мутацией VHL абсолютно показаны аудиологический осмотр, а также КТ или МРТ внутреннего уха с высокой разрешающей способностью, так как раннее хирургическое вмешательство способно сохранить слух. У пациентов с двусторонними опухолями, приводящими к глухоте, слух может быть восстановлен кохлеарным имплантом [108].
У 90% носителей мутации VHL к 60-летнему возрасту имеются те или иные клинические проявления синдрома [45]. На долгосрочный прогноз и смертность обычно влияет наличие гемангиобластом сетчатки и ЦНС, а также карциномы почки на поздних стадиях [31—33]. Таким образом, своевременное обследование и выявление патологии, ассоциированной с VHL-синдромом, является залогом успешного лечения и увеличения продолжительности жизни пациента (табл. 4).
ИСТИНА
Болезнь Гиппеля-Линдау (Von Hippel-Lindau disease) доклад на конференции
- Авторы: Симонян А.С., Сперанская Е.В., Есин А.И., Новикова А.Ю., Петров Р.В., Аджемян Н.А., Егоров В.И.
- Всероссийская Конференция : III-я Научно-Практическая конференция «Актуальные вопросы современной Неврологии»
- Даты проведения конференции: 12 октября 2017
- Дата доклада: 12 октября 2017
- Тип доклада: Устный
- Докладчик: не указан
- Место проведения: Москва, Россия
- Аннотация доклада:
Болезнь Гиппеля-Линдау (VHL) классифицируется как одно из более 7000 редких заболеваний, выявленных на сегодняшний день. В 1927г Шведский патологоанатом Арвид Линдау впервые описал гемангиобластомы в головном и в спинном мозге. Данный научный труд привлек внимание одного из основоположников современной Нейрохирургии – Харви Уильямса Кушинга, который предложил назвать заболевание в честь Арвида Линдау. Долгое время заболевание именовалось в честь Арвида Линдау, но в 1964г научное мировое сообщество обратило внимание на труды Немецкого офтальмолога Эжена фон Гиппеля в которых в 1904г он впервые описал гемангибластомы глаз. Оба ученых в своих работах описывали разные проявления одного и того же патологического состояния, и сегодня с учетом заслуг Эжена фон Гиппеля и Арвида Линдау данная нозология именуется «болезнью Гиппеля - Линдау» В 20% случаев заболевание носит спорадический характер (Мозаичный кариотип - как правило следствие мутаций на ранних стадиях эмбриогенеза). У некоторых пациентов данной группы генетическое тестирование не выявило мутаций в половых клетках, несмотря на то, что в соматических клетках мутации были подтверждены и как следствие у данных людей рождается здоровое потомство. Нельзя исключить также появление подобных случаев вследствие мозаицизма гонад у родителей (Как правило, в результате мутаций на поздних стадиях эмбриогенеза). У данных пациентов вероятность передачи мутантного гена потомству составляет 50% Ген VHL расположен в 3-й хромосом (регион 3p25.3.), содержит 213 аминокислот и способствует выроботке белка pVHL, который является в целом ингибитором опухолевого роста (Ингибитором ангиогенеза). Белок pHVL взаимодействует с 3-мя белками: Elongin B, Elongin C, Cullin 2 (CUL2) и играет ключевую роль в инактивации HIF-1a. Также pVHL снижает продукцию таких факторов ангиогенеза как VEGF. Мутации в гене VHL приводят к нарушению синтеза данного белка, и отсутствие его ингибирующего влияния запускает каскад патологических процессов, что в итоге приводит к различным соматическим проявлениям. Частота заболеваемости VHL одинакова среди женского и мужского населения. Манифестация заболевания может происходить практически в любом возрасте. Заболевание может манифестировать с поражения любого из «органов мишеней». Следует отметить что VHL длительное время может протекать бессимптомно и время постановки диагноза не может считаться временем манифестации у большинства пациентов. Клиническая картина заболевания зависит от количества и степени вовлеченности «органов мишеней», и зачастую сильно варьирует даже у пациентов из одной семьи. Диагностические мероприятия при подазрении на VHL Генетическое тестирование Нейровизуализация (МРТ головного мозга, в том числе с фокусировкой внимания на область внутреннего уха, МРТ шейного, грудного, пояснично-крестцового отделов позвоночника) Флюоресцентная ангиография, офтальмоскопия (Можно также провести дополнительные методы обследования: УЗИ, ЦДК, ОКТ) КТ брюшной полости с контрастным усилением Тест на фракционированные метанефрины, в частности выявление свободного норметанефрина в плазме крови или в анализе суточной мочи. Патогенетического лечения болезни Гиппеля-Линдау на данный момент нет. Наблюдением и лечением пациентов должна заниматься мультидисциплинарная команда специалистов, со знанием особенностей течения данного заболевания.
Болезнь Гиппеля–Линдау (цереброретинальный ангиоматоз)
Болезнь Гиппеля–Линдау – это редкое наследственное нейрокутанное нарушение, которое характеризуется образованием доброкачественных и злокачественных опухолей в нескольких органах. Диагноз ставится на основании клинических критериев и/или молекулярно-генетического тестирования. Лечение - хирургическое или иногда показана радиотерапия или, при ангиомах сетчатки, лазерная коагуляция или криотерапия.
Это заболевание встречается у 1 из 36000 человек и наследуется по аутосомно-доминантному типу Аутосомно-доминантные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения с переменной пенетрантностью. Ген VHL - ген-супрессор опухоли, расположенный на коротком плече хромосомы 3 (3p25.3). У больных с болезнью Гиппеля-Линдау было выявлено более 1500 различных мутаций в этом гене. У 20% людей с этим заболеванием, аномальный ген появился с новыми мутациями.
Болезнь Гиппеля–Линдау чаще всего вызывает
Ангиомы сетчатки глаза
Обычно первые признаки появляются в возрасте 10-30 лет, но могут появиться и раньше.
Симптомы и признаки синдрома VHL
Симптомы болезни фон Гиппеля–Линдау зависят от размера и расположения опухолей. Симптомы могут включать головную боль, головокружение, слабость, атаксию, нарушения зрения и повышенное артериальное давление.
Ангиомы сетчатки, обнаруживаемые при прямой офтальмоскопии, проявляются в виде расширенных артерий, ведущих от диска к периферической опухоли с заполненными кровью венами. Эти ангиомы обычно бессимптомные, но если они расположены в центре и увеличены, они могут привести к существенной потере зрения. Эти опухоли увеличивают риск отслойки сетчатки, отека макулы и глаукомы.
Болезнь Гиппеля – Линдау без лечения может привести к слепоте, повреждению мозга или смерти. Смерть обычно возникает в результате осложнений гемангиобластомы мозжечка или почечно-клеточного рака.
Диагностика болезни Гиппеля-Линдау (VHL)
Визуализационные исследования центральной нервной системы, как правило, МРТ
Иногда молекулярно-генетическое тестирование
Болезнь Гиппеля-Линдау диагностируется при наличии одного из следующих критериев:
Семейный анамнез ВГЛ и наличие ≥ 1 опухоли ВГЛ (гемангиобластома сетчатки, мозга или спинного мозга; феохромоцитома; почечно-клеточная карцинома или эндокринная опухоль поджелудочной железы)
Две или более опухолей, характерных для болезни фон Гиппеля-Линдау (ФГЛ) у пациентов без семейного анамнеза ФГЛ
Если клинические признаки не являются убедительными, диагноз также может быть установлен с помощью молекулярно-генетического тестирования для определения мутаций в гене VHL.
Если у пациента идентифицирована специфическая мутация гена VHL, должно быть проведено генетическое тестирование для определения наличия этой мутации у членов семьи, входящих в группу риска.
Лечение болезни фон Гиппеля-Линдау (VHL-синдрома)
Хирургическое вмешательство, иногда лучевая терапия
При ангиомах сетчатки, лазерная коагуляция или криотерапия
Лечение болезни Гиппеля-Линдау часто включает хирургическое удаление опухоли до того, как она станет опасной. Феохромоцитомы удаляются хирургическим путем, иногда при этом необходимо длительное лечение гипертонии Лечение Артериальная гипертензия – это стойкое повышение систолического артериального давления в покое (≥ 130 мм рт.ст.) и/или диастолического артериального давления (≥ 80 мм рт.ст.). Повышение АД без. Прочитайте дополнительные сведенияДля применения у взрослых пациентов с почечно-клеточным раком, гемангиобластомами центральной нервной системы или эндокринными опухолями поджелудочной железы, которые не требуют немедленного хирургического удаления, в настоящее время доступен бельзутифан, пероральный ингибитор индуцируемого гипоксией фактора 2-альфа. Рекомендуемая доза составляет 120 мг перорально 1 раз в день. Этот препарат можно использовать до начала прогрессирования заболевания или до появления неприемлемой токсичности.
Как правило, чтобы сохранить зрение, ангиомы сетчатки лечат с помощью лазерной коагуляции или криотерапии.
Изучается возможность использования пропранолола для уменьшения размеров гемангиом.
Скрининг с целью проверки осложнений и раннее лечение могут улучшить прогноз.
Скрининг на осложнения
Если диагностические критерии соответствуют болезни Гиппеля-Ландау, пациенты должны регулярно проходить обследование, чтобы выявить осложнения болезни Гиппеля-Ландау, поскольку раннее обнаружение является ключом к предотвращению серьезных осложнений.
Ежегодный сбор анамнеза и физикальное обследование
Ежегодный осмотр глазного дна, начиная с младенчества, на скрининг гемангиобластом сетчатки
Ежегодный мониторинг артериального давления, начиная с 2-летнего возраста, на скрининг феохромоцитом
Ежегодное измерение фракционированных метанефринов в моче или плазме, начиная с 5-летнего возраста с целью скрининга феохромоцитом
МРТ головного и спинного мозга каждые 2 года, начиная с возраста 11 лет, для скрининга на гемангиобластомы центральной нервной системы
Аудиография каждые 2 года, начиная с 11 лет, на скрининг опухолей эндолимфатического мешка
МРТ брюшной полости или УЗИ каждые 2 года, начиная с 15-летнего возраста, для скрининга на почечно-клеточный рак, феохромоцитомы и опухоли поджелудочной железы
Лица, не соответствующие диагностическим критериям БГЛ, но имеющие гаметную мутацию, или которые не были тестированы, но являются членами семьи 1-й и 2-й линии родства пациента, страдающего БГЛ, должны быть также обследованы с помощью следующих методов:
Ежегодное обследование с целью выявления неврологических симптомов, нарушений зрения и слуха
Ежегодное офтальмологическое обследование для выявления нистагма, косоглазия и белых зрачков
Ежегодный мониторинг артериального давления
Справочные материалы по скринингу
1. Maher ER: Von Hippel-Lindau disease. Curr Mol Med 4(8):833–842, 2004. doi: 10.2174/1566524043359827
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Синдром фон Гиппеля-Линдау на МРТ
- Главная /
- Редкие болезни /
- Энциклопедия заболеваний /
- Болезнь Гиппеля Линдау
Болезнь Гиппеля-Линдау ( VHL ), Синдром Гиппеля-Линдау
Von Hippel–Lindau disease , Von Hippel–Lindau syndrome
OMIM – 193300 МКБ10 – Q 85.8
Болезнь Гиппеля-Линдау классифицируется как одно из более 7000 редких заболеваний, выявленных на сегодняшний день. Частота заболеваемости среди населения 1/25000-50000. VHL причисляется к наследственным (Генетическим) заболеваниям и наследуется аутосомно-доминантным путем (~80% случаев).

В 20% случаев заболевание носит спорадический характер (Мозаичный кариотип - как правило следствие мутаций на ранних стадиях эмбриогенеза). У некоторых пациентов данной группы генетическое тестирование не выявило мутаций в половых клетках, несмотря на то, что в соматических клетках мутации были подтверждены и как следствие у данных людей рождается здоровое потомство. Нельзя исключить также появление подобных случаев вследствие мозаицизма гонад у родителей (Как правило, в результате мутаций на поздних стадиях эмбриогенеза). У данных пациентов вероятность передачи мутантного гена потомству составляет 50%.
Генетика: Ген VHL расположен в 3-й хромосом (регион 3 p 25.3.), содержит 213 аминокислот и способствует выроботке белка pVHL , который является в целом ингибитором опухолевого роста (Ингибитором ангиогенеза).

Белок pHVL взаимодействует с 3-мя белками: Elongin B , Elongin C , Cullin 2 ( CUL 2) и играет ключевую роль в инактивации HIF -1 a . Также pVHL снижает продукцию таких факторов ангиогенеза как VEGF . Мутации в гене VHL приводят к нарушению синтеза данного белка, и отсутствие его ингибирующего влияния запускает каскад патологических процессов, что в итоге приводит к различным соматическим проявлениям.
Клинические проявления: Основными проявлениями заболевания являются
- Гемангиобластомы мозжечка, ствола головного мозга, позвоночного канала,
- Гемангиобластомы сетчатки,
- Новообразования эндолимфатичесого мешочка внутреннего уха,
- Гемангиомы печени, Аденомы печени,
- Феохромоцитомы или параганглиомы,
- Кисты поджелудочной железы, цистаденомы или нейроэндокринные опухоли,
- Кисты почек, почечно-клеточная карцинома,
- Цистаденомы придатка яичка (У мужчин), Папиллярный рак широкой связки (У женщин),
- Доброкачественные изменения в легких.

Клиническая картина заболевания зависит от количества и степени вовлеченности «органов мишеней», и зачастую сильно варьирует даже у пациентов из одной семьи.
Манифестация: Манифестация заболевания может происходить практически в любом возрасте. Заболевание может начаться с поражения любого из «органов мишеней». Следует отметить, что VHL длительное время может протекать бессимптомно и время постановки диагноза не может считаться временем манифестации у большинства пациентов.
Типы заболевания: Различают несколько вариантов заболевания, которые протекают с вовлечением органов мишеней в различных комбинациях.

Диагностические мероприятия при подозрении на VHL
- Генетическое тестирование
- Нейровизуализация (МРТ головного мозга, в том числе с фокусировкой внимания на область внутреннего уха, МРТ шейного, грудного, пояснично-крестцового отделов позвоночника)
- Флюоресцентная ангиография, офтальмоскопия (Можно также провести дополнительные методы обследования: УЗИ, ЦДК, ОКТ)
- КТ брюшной полости с контрастным усилением
- Тест на фракционированные метанефрины, в частности выявление свободного норметанефрина в плазме крови или в анализе суточной мочи.
Лечение: Патогенетического лечения болезни Гиппеля-Линдау на данный момент нет. Наблюдением и лечением пациентов должна заниматься мультидисциплинарная команда специалистов, со знанием особенностей течения данного заболевания.
- Тактика лечения гемангиобластом головного мозга
- Хирургическое лечение
- Радиохирургия ( Cyber Knife )
- Наблюдение
- Тактика лечения гемангиобластом сетчатки
- Основным методом лечения является транспупиллярная лазерная коагуляция ангиоматозного узла/капиллярной гемангиомы (Эффективность высока при размерах опухоли до 3-х мм).
- Также применяются - Криотерапия, брахитерапия, наружная лучевая терапия, транпупиллярная термотерапия, фотодинамическая терапия
- В случае развития осложнений применяется (Отслойка сетчатки) - Витроретинальная хирургия.
- Тактика лечения поражений брюшной полости
- Кисты и цистаденомы поджелудочной железы обычно не требуют хирургического вмешательства
- PNET : Одним из возможных методов лечения является хирургический, В настоящий момент идет пересмотр рекомендация по лечению PNET .
- Почечно-клеточная карцинома: Основной метод лечения хирургический, возможно применение радиочастотной абляции и криохирургии.
- Феохромоцитомы: Хирургическое лечение ( Laparoscopic partial adrenalectomy ) с дальнейшим подбором консервативной терапии.
Образ жизни: Важное значение для качества жизни пациентов имеют - питание, физическая активность, эмоциональное состояние.


Наблюдение пациентов: осуществляется мультидисциплинарной командой, в зависимости от проявлений заболевания. Координируют деятельность команды
Болезнь Гиппеля-Линдау: гемангиобластома сетчатки
Информация только для специалистов в сфере медицины, фармации и здравоохранения!

Содержание
- Гемангиобластома центральной нервной системы
- Гемангиобластома сетчатки
- Светлоклеточный почечно-клеточный рак
- Феохромоцитома
- Опухоли эндолимфатического мешка среднего уха
- Серозная цистаденома и нейроэндокринные опухоли поджелудочной железы
- Папиллярная цистаденома придатка яичка и широкой связки матки
Молекулярная биология болезни Гиппеля-Линдау

Ген фон Гиппеля-Линдау (VHL) был картирован на хромосоме 3p25 и клонирован в начале 1990-х, а дальнейшие исследования в понимании функции гена VHL продолжались в течение следующих 20 лет. Продукт этого гена, белок VHL, действует как супрессор опухолей. Как и в случае с патогенными вариантами некоторых других генов-супрессоров опухолей (например, гена ретинобластомы 1, RB1), модель «двух ударов» была подтверждена для БГЛ, при которой вариант потери функции зародышевой линии инактивирует одну копию VHL-гена во всех клетках. Для развития опухолей, связанных с VHL, должна произойти потеря экспрессии второго нормального аллеля либо из-за соматических изменений, либо из-за делеции второго аллеля, либо из-за гиперметилирования его промотора. При спорадическом почечно-клеточном раке инактивация VHL за счет соматических изменений обоих аллелей очень распространена.
За последние два десятилетия были достигнуты значительные успехи в понимании биологии, лежащей в основе формирования опухолей, связанных с VHL. Белок VHL образует стабильный комплекс с несколькими другими белками, включая элонгины В и С, а также куллин 2. Этот комплекс нацелен на несколько белков для протеасомной деградации, тем самым регулируя их уровни внутри клетки. Компонент VHL этого комплекса функционирует как убиквитинлигаза E3 для молекул-мишеней. После связывания с комплексом VHL молекулы-мишени ковалентно связываются с убиквитином, облегчая деградацию протеасомой.
В дополнение к своей функции убиквитинлигазы E3, VHL выполняет несколько других важных клеточных функций, включая поддержание первичной реснички, регуляцию цитокинеза, контроль функции микротрубочек, целостность внеклеточного матрикса и регуляцию клеточного цикла. Патогенные варианты гена VHL также связаны с врожденными формами истинной полицитемии.
Клиническая картина капиллярной гемангиобластомы сетчатки

Капиллярные гемангиобластомы сетчатки обычно локализуются либо в периферической части сетчатки, либо в юкстапапиллярной области. Потеря зрения при капиллярных гемангиобластомах сетчатки обычно вызывается экссудацией из опухоли, вызывающей отек сетчатки, или тракционными эффектами, при которых глиальная пролиферация на поверхности опухоли вызывает стрии и деформацию сетчатки. Кроме того, капиллярные гемангиобластомы сетчатки могут кровоизлиять, что приводит к отслойке сетчатки, глаукоме и потере зрения.
Капиллярные гемангиобластомы сетчатки обнаруживаются у 70% пациентов с БГЛ к возрасту 60 лет; они часто многоочаговые и двусторонние. По сравнению с пациентами со спорадическими гемангиобластомами сетчатки пациенты с БГЛ намного моложе и чаще имеют множественные поражения. В одной серии из 31 пациента с БГЛ и 37 пациентов без неё пациенты с БГЛ были моложе (18 лет против 36 лет соответственно), имели в среднем четыре опухоли и были более склонны к развитию новых опухолей, чем пациенты без заболевания. заболевание.
В исследовании 890 пациентов с БГЛ у 335 пациентов была капиллярная гемангиобластома сетчатки как минимум в одном глазу. Поражения были обнаружены односторонне у 42% и двусторонне у 58% пострадавших пациентов. Корреляции между возрастом, полом и латеральностью поражения выявлено не было. Из вовлеченных глаз 87% имели опухоли, которые можно было визуализировать индивидуально; из них опухоли обычно обнаруживались только в периферической части сетчатки (85%) и реже в юкстапапиллярной области (15%). Количество опухолей на периферии в среднем составляло 2,5+/-1,8 на глаз, при этом 25% глаз имели поражение более одного квадранта сетчатки. Оценка взаимосвязи генотип-фенотип при капиллярной гемангиобластоме сетчатки показала, что у 15% людей с вариантами, которые приводят к полной потере белка VHL, развилась гемангиобластома по сравнению с общей распространенностью в популяции пациентов 37%. Было обнаружено, что риск потери зрения увеличивается с возрастом, хотя количество опухолей существенно не увеличивается в зависимости от возраста.
Как и в случае с кажущимися спорадическими гемангиобластомами центральной нервной системы (ЦНС), любой пациент с капиллярной гемангиобластомой сетчатки (особенно в возрасте до 40 лет) должен пройти генетическое тестирование зародышевой линии на наличие патогенных вариантов гена VHL.
Рутинное наблюдение за капиллярной гемангиобластомой сетчатки рекомендуется для пациентов с заболеванием VHL из-за его высокой частоты. Частое появление таких поражений в детстве делает важным инициировать офтальмологическое наблюдение в педиатрической популяции после постановки диагноза, и это является одной из причин, по которой рекомендуется генетическое тестирование на патогенные варианты гена VHL у детей раннего возраста.
Диагностика капиллярной гемангиобластомы сетчатки

Диагноз болезни (фон) Гиппеля-Линдау (БГЛ) обычно устанавливается путем обнаружения зародышевого патогенного (обычно с потерей функции) варианта гена VHL. Это чаще всего наблюдается у пациентов, которые проходят генетическое тестирование после того, как у них диагностировано одно проявление БГЛ, или у тех, кто проходит тестирование, потому что у их близких родственников диагностирована эта болезнь. Диагноз также может возникнуть, когда генетическое тестирование проводится по другой причине и неожиданно обнаруживает вторичный патогенный вариант VHL. В редких случаях у пациентов, у которых нет доступа к генетическому тестированию, диагноз болезни может быть основан на клинических критериях (например, у пациентов с одним поражением, связанным с БГЛ, и соответствующим семейным анамнезом, или у пациентов с множественными поражениями, связанными с БГЛ).
Пациентов с подозрением на БГЛ следует направлять в специализированные центры для оценки, генетического консультирования и окончательного диагноза с помощью генетического тестирования, даже если в семейном анамнезе нет этой болезни.
Пациентов следует направлять на соответствующую генетическую консультацию в сочетании с генетическим тестированием на патогенные варианты гена VHL. Болезнь наследуется по аутосомно-доминантному типу, и больные люди имеют 50-процентную вероятность передачи ассоциированного с болезнью варианта гена VHL каждому потомку. Учитывая разный возраст появления опухоли, большинство людей с БГЛ доживают до зрелого возраста и рожают детей в основном до постановки диагноза. Таким образом, нет ничего необычного в том, чтобы увидеть многопоколенческие родословные БГЛ со многими пораженными людьми, каждый из которых имеет несколько разные модели диагнозов опухолей и разный возраст начала заболевания.
Среди редких пациентов с соматическим мозаицизмом риск для потомства зависит от того, несет ли зародышевая ткань патогенный вариант, хотя это обычно не определяется клинически. Таким образом, пациенты с документально подтвержденным мозаицизмом должны быть проинформированы о том, что риск рождения больного ребенка может достигать 50% и что любой больной ребенок унаследует патогенный вариант в 100% своих клеток и потенциально будет иметь более тяжелые проявления болезни, чем мозаичный родитель.
Диагноз БГЛ у ребенка здоровых родителей может быть очень тревожным, и следует тщательно объяснить концепцию патогенных вариантов de novo или переменной экспрессивности (например, когда у родителя еще не была диагностирована опухоль, связанная с БГЛ). Никогда не следует предполагать, что здоровые родители отрицательны на патологический вариант гена VHL без прямого генетического тестирования. Родителей следует успокоить, а возможную вину смягчить, объяснив, что патогенный вариант de novo вряд ли может быть результатом какого-либо действия, имевшего место непосредственно перед беременностью или во время нее.
Растет обеспокоенность родителей относительно того, когда предоставлять информацию о диагнозе детям с положительным генетическим тестом на VHL. В общем, лучше всего передавать эту информацию в различных условиях по мере взросления ребенка, и родители могут получить помощь от медицинского работника при инициировании этих дискуссий.
Лечение капиллярной гемангиобластомы сетчатки
Лечение капиллярной гемангиобластомы сетчатки требует, чтобы преимущества лечения были сбалансированы с потенциальными осложнениями, связанными с лечением. Данные о том, можно ли внимательно наблюдать за небольшими поражениями без специфического лечения, до тех пор, пока не появятся какие-либо признаки роста или симптомы, противоречивы. Некоторые группы рекомендуют лечить капиллярную гемангиобластому сетчатки сразу после обнаружения (чтобы предотвратить рост и осложнения), в то время как другие ждут некоторого изменения в размере, прежде чем начинать лечение. Для тех, кто начинает лечение, мы предлагаем лазерную фотокоагуляцию, а не другие методы лечения. Другие альтернативные варианты включают фотодинамическую терапию или лучевую терапию. Системная терапия белзутифаном является приемлемым вариантом для пациентов, которым противопоказана местная терапия из-за близости опухоли к зрительному нерву или множественных прогрессирующих поражений. Клинические испытания приветствуются, если таковые имеются.
Местная терапия. Лазерная фотокоагуляция эффективна более чем в 70% случаев, как правило, при однократном лечении и является предпочтительным методом лечения. Исключением является то, что гемангиобластомы зрительного нерва не следует лечить этими методами из-за вредных побочных эффектов на нормальную сетчатку. Фотодинамическая терапия также может рассматриваться как вариант лечения капиллярной гемангиобластомы сетчатки, хотя данные о ее эффективности ограничены. Внешняя лучевая терапия может быть использована в качестве терапии спасения, если другие методы оказались неэффективными.
Белзутифан, ингибитор индуцируемого гипоксией фактора-2альфа (HIF-2альфа), является вариантом лечения пациентов с капиллярными гемангиобластомами сетчатки, расположенными близко к зрительному нерву, или пациентов с множественными прогрессирующими гемангиобластомами; такие пациенты, как правило, не подходят для местной терапии. В исследовании фазы II белзутифан улучшил состояние во всех 16 глазах (100%) у 12 пациентов с подлежащими оценке гемангиобластомами сетчатки.
Экспериментальная терапия (антиангиогенные средства) – необходимы дальнейшие исследовательские исследования для лучшего понимания биологии исходной гемангиобластомной клетки и ее эндотелия, а также для разработки активной системной терапии. Несколько ингибиторов рецепторов фактора роста эндотелия сосудов (VEGF), которые препятствуют ангиогенезу, такие как сунитиниб, пазопаниб, бевацизумаб и ранибизумаб, продемонстрировали ограниченную эффективность при гемангиобластоме сетчатки.
Наблюдение за пациентами с капиллярной гемангиобластомы сетчатки

Заболеваемость и смертность у пациентов с болезнью (фон) Гиппеля-Линдау (БГЛ) значительно снизились благодаря лучшему пониманию естественного течения серьезных клинических проявлений заболевания, более совершенным методам визуализации и улучшениям в терапии. Наблюдение важно не только для выявления новых поражений на ранней стадии, но и для наблюдения за небольшими бессимптомными поражениями на предмет признаков прогрессирования.
Протоколы наблюдения сосредоточены на гемангиобластомах (включая капиллярные гемангиобластомы сетчатки), почечно-клеточном раке (ПКР), феохромоцитомах и аудиологическом исследовании, учитывая повышенный риск опухолей эндолимфатического мешка (ЭЛСТ) у пациентов с БГЛ. Рекомендации по наблюдению могут быть адаптированы к каждому пациенту с учетом текущего или предшествующего диагноза опухоли. Тем не менее, все люди с БГЛ, даже если в настоящее время они бессимптомны, должны понимать, что у них могут развиться проявления болезни, и им будет полезно следовать рекомендациям по эпиднадзору.
Несколько организаций предоставляют обновленные руководства по эпиднадзору, учитывающие современные методы визуализации и лабораторной диагностики. Международная группа клиницистов, занимающихся лечением детей с БГЛ, была созвана в 2016 году в рамках семинара Американской ассоциации исследований рака (AACR) по детской предрасположенности к раку. Группа рассмотрела как американские, так и европейские схемы по БГЛ и опубликовала рекомендации по эпиднадзору, которые предусматривали повышение интенсивности и более раннее начало скрининга. Эти рекомендации впоследствии были оценены консенсусной комиссией, сформированной Альянсом БГЛ, состоящей из клиницистов, занимающихся всеми областями знаний, связанных с лечением болезни, и представителей семинара AACR. Ниже приводится сводка этих рекомендаций.
Читайте также: