Синдром Элерса-Данлоса
Обновлено: 27.04.2024
РНЦХ им. акад. Б.В. Петровского РАМН
РНЦХ им. акад. Б.В. Петровского РАМН, Москва
Отделение кардиохирургическое Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
лаборатория медицинской генетики Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
лаборатория медицинской генетики Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
ФГБУ РНЦХ им. акад. Б.В. Петровского РАМН
РНЦХ им. акад. Б.В. Петровского РАМН, Москва
Диагностика и хирургическое лечение больных с cосудистым типом синдрома Элерса—Данло
Журнал: Кардиология и сердечно-сосудистая хирургия. 2017;10(6): 79‑84
РНЦХ им. акад. Б.В. Петровского РАМН
РНЦХ им. акад. Б.В. Петровского РАМН
РНЦХ им. акад. Б.В. Петровского РАМН, Москва
Отделение кардиохирургическое Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
лаборатория медицинской генетики Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
лаборатория медицинской генетики Российского научного центра хирургии им. акад. Б.В. Петровского, Москва, Россия
ФГБУ РНЦХ им. акад. Б.В. Петровского РАМН
РНЦХ им. акад. Б.В. Петровского РАМН, Москва
Чрезвычайные ситуации в сосудистой хирургии, связанные со спонтанными разрывами (СР) периферических артерий крупного и среднего диаметра, опасны для жизни и требуют своевременного неотложного лечения [1, 2]. Исследование Global Burden Disease [3, 4] продемонстрировало, что общее число летальных исходов по причине аневризм и расслоения аорты выросло с 2,49 на 100 000 до 2,78 на 100 000 случаев за период с 1990 по 2010 г.
Пациенты с наследственными дисплазиями соединительной ткани (НДСТ) в течение жизни могут неоднократно нуждаться в разных типах хирургических вмешательств, в первую очередь в реконструктивных вмешательствах на сосудах и сердце. НДСТ — клинически и генетически гетерогенная группа заболеваний, связанная с наследственными нарушениями синтеза и функционирования коллагеновых и эластических белков. Клиническая дифференциальная диагностика НДСТ чрезвычайно трудна вследствие сходства симптоматики. И если «марфаноидный фенотип» хорошо известен кардиологам и сосудистым хирургам, то внешний вид пациентов с сосудистым типом синдрома Элерса—Данло (СЭД IV) редко дифференцируется в клинике. Клиническая диагностика синдрома СЭД основана на Вильфраншских критериях 1998 г. [9]. Сбор семейного анамнеза и проведение ДНК-диагностики является необходимым этапом в подтверждении диагноза сосудистого типа СЭД [27]. Лабораторное тестирование рекомендуется лицам, имеющим два и более диагностических симптома. Верификация генетической причины заболевания у пробанда позволяет проводить подтверждающую, раннюю и пресимптоматическую диагностику заболевания (в том числе пренатальную по запросу семьи) у всех родственников, доступных для обследования [16]. Заболевание является полиорганным и жизнеугрожающим, вследствие риска разрыва сосудов среднего и крупного калибра, а также полых органов (кишечник, матка, мочевой пузырь). Продолжительность жизни больных значительно снижена, при естественном течении заболевания выживаемость к 50 годам не превышает 50% [5, 6]. Частота СЭД IV (MIM 130 050) составляет 1:250 000—1:100 000 населения, однако возможна недооценка истинной распространенности этого заболевания вследствие гиподиагностики [5].
В отечественной литературе мы не обнаружили случаев хирургического лечения пациентов с генетически подтвержденным СЭД IV типа [1, 2, 11]. В настоящей работе представлен пример мультидисциплинарного подхода, обеспечившего успешное лечение 2 пациентов с сочетанием хирургического лечения, таргетной терапии, а также медико-генетическим консультированием и ДНК-диагностикой пациентов с сосудистым типом СЭД.
Материал и методы
В отделении кардиохирургии № 1 (хирургии аорты и ее ветвей) Российского научного центра хирургии им. Б.В. Петровского за последние 10 лет было только 2 пациента с генетически подтвержденным СЭД IV типа, что составило 0,2% от всех больных с различными формами дисплазий, пролеченных за этот период. Всех пациентов с подозрением на НДСТ, госпитализированных в отделение, консультировали в лаборатории медицинской генетики ФГБНУ «Российский научный центр хирургии им. акад. Б.В. Петровского», что позволяло уточнить диагноз и корригировать дальнейшее лечение.
Клинические данные пациентов приведены в таблице. Таблица 1. Клинические характеристики пациентов Примечание. ЛПклА — левая подключичная артерия; ПА — почечная артерия; ВоА — восходящая аорта; АК — аортальный клапан; СР миокарда — спонтанный разрыв миокарда.
Больной З., 16 лет, поступил в ноябре 2010 г. для оперативного лечения в отделение хирургии аорты и ее ветвей РНЦХ им. акад. Б.В. Петровского.
Из анамнеза известно, что в ноябре 2008 г. почувствовал нарастающую боль в левом плечевом суставе, позже появилась пульсирующая припухлость. Госпитализирован бригадой СМП в больницу Сыктывкара, где диагностирован разрыв аневризмы левой подключичной артерии, с прорывом в левую плевральную полость, гемоторакс объемом до 2 л. Экстренно выполнено протезирование левой подключичной артерии. Через 6 мес после вмешательства при проведении планового обследования выявлен тромбоз протеза с признаками ишемии левой верхней конечности. В апреле 2010 г. по месту жительства в отделении микрохирургии предпринята попытка репротезирования, однако операция оказалась безуспешной ввиду выраженных изменений стенки подключичной артерии (резкое истончение, отсутствие эластичности). При гистологическом исследовании отмечалась очаговая аплазия мышечного слоя и отсутствие внутренней эластической мембраны артерии. Тогда же, по данным ангиографии, диагностирована аневризма правой почечной артерии.
При поступлении предьявлял жалобы на слабость и сильные боли в левой верхней конечности. Больной истощен (рост 170 см, масса тела 45 кг). Отмечается выраженная термоасимметрия верхних конечностей, а также гипотрофия, снижение силы и тонуса в левой верхней конечности.

Больной проконсультирован врачом-генетиком. При осмотре были выявлены признаки дисплазии соединительной ткани (рис. 1). Рис. 1. Особенности фенотипа, характерные для сосудистого типа СЭД. а — гипермобильность суставов кистей, б — «папиросные рубцы» после травмы.
Кожа гиперэластичная, тонкая, подкожные вены ясно видны под кожей грудной клетки, несколько «папиросных» рубцов, послеоперационные келоидные рубцы, гипотония лицевых мышц, выраженная гипермобильность суставов, искривление позвоночника, небольшое воронкообразное искривление грудины, плоскостопие обеих стоп, синдактилия II—IV пальцев стоп, небо высокое, неправильный рост зубов, в анамнезе пупочная и паховая грыжи. Отмечал частое появление подкожных гематом, усилившихся в последнее время. Указаний на заболевания печени, которые могли бы послужить причиной гипокоагуляции, в анамнезе или истории болезни не обнаружено. Характерный для сосудистого типа СЭД фенотип (узкий нос, тонкие губы), обусловленный снижением подкожного жирового слоя, не отмечался [5, 10]. Семейный анамнез не известен (ребенок усыновлен).

С целью подтверждения диагноза, последующего медико-генетического консультирования семьи и оценки риска хирургических осложнений был выполнен поиск мутаций в гене COL3A1 методом прямого секвенирования по Сенгеру, кодирующих участков и прилегающих интронных областей. Была выявлена замена p. Gly183Ser в 6-м экзоне гена COL3A1 в гетерозиготном состоянии, приведшая к появлению миссенс-мутации (рис. 2). Рис. 2. ДНК-диагностика сосудистого типа синдрома СЭД у пациента З. Фрагмент прямого секвенирования 6-го экзона гена COL3A1. Стрелкой указана мутация c.547G>A (p.Gly183Ser) в гетерозиготном состоянии.

Пациенту выполнено аутовенозное сонно-плечевое шунтирование слева. Вследствие выраженных изменений больших подкожных вен на обеих нижних конечностях (малый диаметр, сегментарная деструкция стенки) шунт сформирован из трех фрагментов, забранных с обеих нижних конечностей. Хирургическое вмешательство усложнилось наличием грубых рубцовых деформаций в этой зоне после двух ранее перенесенных реконструкций (рис. 3, а,). Рис. 3. Интраоперационное фото: а — послеоперационный рубец в месте предыдущих реконструкций; б — дистальный анастомоз с плечевой артерией; в — проксимальный анастомоз с левой общей сонной артерией; г — конечный вид реконструкции из надключичного доступа. Левая общая сонная артерия выделена из надключичного доступа, левая плечевая артерия эксплорирована в верхней трети плеча. Выполнен анастомоз аутовенозного шунта с плечевой артерией по типу конец в бок атравматической полипропиленовой нитью 6/0. Шунт проведен под малой и большой грудными мышцами и над ключицей к левой общей сонной артерии. Сформирован анастомоз шунта с левой общей сонной артерией по типу конец в бок атравматической полипропиленовой нитью 6/0 (см. рис. 3, б, в).
Послеоперационный период протекал без осложнений. При контрольном дуплексном сканировании сонно-плечевой аутовенозный шунт слева проходим, кровоток по шунту Vs=2,8 м/с. Больной выписан на 7-е сутки после операции. Протезирование почечной артерии по поводу аневризмы выполнено в дальнейшем по месту жительства.
Больной П., 46 лет, поступил для оперативного лечения в отделение хирургии аорты и ее ветвей РНЦХ им. акад. Б.В. Петровского с жалобами на периодические боли в области сердца, давящего характера, возникающие при физической нагрузке, учащенное сердцебиение.
Из анамнеза: три сотрясения головного мозга, ишемический инсульт, перевязка правой внутренней подвздошной артерии по причине разрыва ложной посттравматической аневризмы верхней ягодичной артерии. Семейный анамнез отягощен: родители умерли от сердечно-сосудистых заболеваний, сестра страдает варикозной болезнью с 15 лет.
При физикальном осмотре отмечаются признаки дисплазии соединительной ткани: кожа гиперэластичная, бархатистая на ощупь, «положительный симптом щипка», характерные потертости на коленях, «папиросные» рубцы, экхимозы, нарушение осанки, поперечное плоскостопие обеих стоп, высокое небо, неправильный рост зубов, выраженная гипермобильность суставов.

Заключение генетика: СЭД. Был выполнен поиск мутаций в гене COL3A1 методом прямого секвенирования по Сенгеру, кодирующих участков и прилегающих интронных областей (рис. 4). Рис. 4. ДНК-диагностика сосудистого типа синдрома СЭД у пациента П. Фрагмент прямого секвенирования 45-го экзона гена COL3A1. Стрелкой указана мутация c. 3301G>A (p.Gly1101Arg) в гетерозиготном состоянии.
По данным эхокардиографии диагностирован порок аортального клапана с аортальной недостаточностью 2—3 степени, кардиомегалия (КДР 6,5 см, КДО 220 мл). При Д.С. периферических артерий выявлена патологическая извитость и деформация хода внутренних сонных артерий с обеих сторон, наружные подвздошные артерии S-образно извиты.
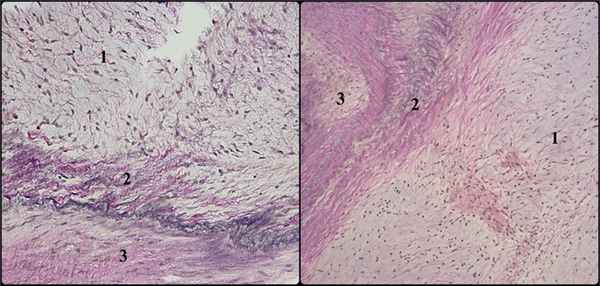
Пациенту выполнено протезирование аортального клапана механическим протезом. Из интраоперационных особенностей отмечается спонтанный разрыв корня аорты на 2/3 диаметра проксимальнее области аортотомного шва ввиду выраженной дисплазии тканей и повторное подключение аппарата И.К. Ткани взяты на гистологическое исследование (рис. 5). Рис. 5. Морфологическая картина дисплазии соединительной ткани аортального клапана и аорты: рыхлая соединительная ткань с явлениями дезорганизации и миксоматозом (1), истончение и неравномерное распределение эластических волокон (2), очаговый склероз (3). Окраска пикрофуксином по Ван-Гизону с докраской на эластические волокна — ×100 (а), × 50 (б).
Произведена пластика восходящей аорты ксеноперикардом с использованием тефлоновых полосок. Ранний послеоперационный период осложнился кровотечением с тампонадой сердца и гемотораксом после удаления дренажей на 2-е сутки. При ревизии перикарда и средостения отмечались спонтанные разрывы в области передней стенки правого желудочка и задней стенки левого желудочка. Раны ушиты на тефлоновых прокладках. Пациент выписан на 12-е сутки.
В течение жизни пациенты, имеющие клинические проявления сосудистого типа СЭД (аневризмы периферических и висцеральных артерий крупного и среднего диаметра, спонтанные разрывы полых органов в анамнезе, варикозную болезнь в молодом возрасте, каротидно-кавернозные фистулы, гипермобильность суставов, гиперэластичнось кожи, стрии, папиросные и келоидные рубцы, деформации грудной клетки, позвоночника, нижних конечностей, патология глаз, грыжи и т. п.), с высокой частотой попадают в хирургический стационар для оказания различных видов хирургической помощи [11]. Зачастую первичное вмешательство является неэффективным, как показано в первом клиническом примере, что связано в первую очередь с системной патологией, и поэтому требуются повторные операции [12]. Однако в отсутствие патогномоничных биомаркеров при стертых и неспецифичных клинических проявлениях клиническая диагностика НДСТ и оценка хирургических рисков только по фенотипическим особенностям очень субъективна. Поэтому «золотым стандартом» диагностики сосудистого типа СЭД является подтверждающая ДНК-диагностика (выявление мутаций в гене COL3A1), которая была впервые разработана в лаборатории медицинской генетики РНЦХ им. акад. Б.В. Петровского.
Коллаген III типа является основной структурной единицей в архитектонике соединительнотканного остова дермы, связок, сухожилий, капсул суставов, стенки аорты и крупных артерий, вен, подслизистого слоя полых органов, что напрямую коррелирует с широким спектром осложнений при IV типе СЭД [17]. Коллаген III типа принадлежит к гомотримерным фибриллярным коллагенам. Он формируется при сочетании трех мономеров или α-цепей белка-предшественника протоколлагена. Последовательность аминокислот тройной спирали характеризуется повторами Gly-X-Y последовательностей, где X и Y часто представлены пролином и гидроксипролином. Для того чтобы обеспечить правильное связывание α-мономеров и стабильность коллагеновых волокон, не должно быть перерывов в повторах глицин-X-Y триплетов, а длина тройной спирали должна оставаться одинаковой для каждой α-цепи. Поэтому практически все мутации, приводящие к замене глицина на другую аминокислоту (как у представленных пациентов), являются патогенными и приводят к снижению содержания коллагена III до 10—15% от нормы [14, 15]. Другие мутации в гене COL3A1, при которых содержание нормального коллагена составляет не менее 50%, ассоциированы с более мягким течением заболевания, более поздними сосудистыми осложнениями, отсутствием кишечных осложнений и лучшим долгосрочным прогнозом [16].
M. Pepin и соавт. [8] на когорте из 220 пациентов также доказали, что в 92% случаев причиной отсроченной смерти у пациентов с СЭД IV типа являлись сосудистые осложнения.
Варикозная болезнь часто встречается у данной категории пациентов, однако хирургическое лечение приводит в большинстве случаев к рецидивам. Разрыв кишечника чаще всего включает сигмовидную кишку.
Каждый пациент требует специализированного анестезиологического подхода [23]. Анестезиологические службы должны быть готовы к переливанию большого объема гемокомпонентов, сердечным аномалиям и порокам развития, требующим определенной оценки во время предоперационной подготовки [24]. Особое внимание стоит уделить нарушениям проводимости миокарда, вторичной митральной недостаточности и атриомегалии. Согласно руководству National Institute for Clinical Excellence, все постановки центральных венозных катетеров и артериальные пункции должны проводиться строго под УЗИ-контролем в целях снижения риска образования гематом, ложных аневризм, спонтанных разрывов сосудов [23]. В литературе описаны случаи эрозии сосудистой стенки в месте стояния катетера, осложненные эффузией крови в плевральную полость и перикард. Ввиду этого катетеры необходимо удалять как можно скорее [25]. Также персонал должен быть ориентирован на максимально прецизионную и бережную интубацию, ввиду склонности больных к кровоточивости и травматизации тканей. Обычная ларингоскопия может стать причиной травмы десен, слизистой полости рта, мягкого неба, вывиха височно-нижнечелюстного сустава. Рекомендовано использовать вспомогательную фиброоптическую бронхоскопию для интубации с целью минимизации травмы и определения точного положения интубационной трубки [26]. Любая фиксация катетеров, трубок, электродов может сопровождаться образованием экхимозов. При укладке больного возможна дислокация суставов. Все внутримышечные и подкожные инъекции могут сопровождаться образованием гематом [23].
Тип генетического повреждения и расположение мутации позволяют адекватно ориентировать врача на этапе планирования вмешательства с целью достижения максимально радикального результата хирургического лечения. Деликатное, атравматичное, прецизионное отношение к тканям, использование зажимов с мягким покрытием, баллонных окклюдеров, мягких ретракторов, тефлоновых прокладок при наложении анастомозов позволяют исключить осложнения в послеоперационном периоде.
В настоящее время разрабатывается новый подход к лечению, направленный на репрессию мутантного гена с использованием специфических аллельспецифичных малых интерферирующих РНК (siRNA). Эффективность такого подхода уже была показана в культуре кожных фибробластов больного с васкулярным типом СЭД, что может служить основой для разработки максимально персонализированной этиологической терапии этого заболевания [15].
Заключение
Подтвержденный диагноз сосудистого типа СЭД, как и почти любого системного заболевания, утяжеляет дальнейший прогноз даже при успешно выполненном хирургическом лечении и требует детального консультирования пациентов перед выпиской. Всем пациентам также рекомендовано динамическое наблюдение с ежегодными консультациями профильных специалистов. Предпочтения в диагностике стоит отдавать неинвазивным методам. Аутосомно-доминантный тип наследования сосудистого типа СЭД позволяет ожидать накопления заболевания в семье, что требует выполнения каскадного семейного скрининга. План динамического наблюдения и профилактики осложнений для родственников-носителей мутации в гене COL3A1 должен соответствовать программе мониторинга здоровья пациента.
Синдром Элерса — Данлоса: что это и как с ним жить

Удивительная гибкость, эластичная кожа и постоянная боль. Все это — проявления синдрома Элерса — Данлоса. Рассказываем о том, можно ли его вылечить и как люди с этим синдромом чувствуют себя в обществе
Что такое синдром Элерса — Данлоса
Синдром Элерса — Данлоса (Синдром Элерса-Данло, СЭД) — это группа наследственных заболеваний, которые поражают соединительные ткани: в первую очередь кожу, суставы и стенки кровеносных сосудов. Задача соединительной ткани — обеспечить прочность и эластичность основных структур тела. Синдром Элерса — Данлоса делает суставы слишком гибкими, а кожу — растяжимой и хрупкой. Это становится проблемой, если, например, образуется рана, на которую нужно наложить швы — кожа у человека с СЭД часто недостаточно прочна, чтобы удержать их. А более тяжелые формы СЭД могут даже привести к разрыву стенок кровеносных сосудов, кишечника или матки.
Вызывает все эти проявления генетическая проблема с выработкой коллагена — важнейшего белка мышц, кровеносных сосудов, костей и других органов. Люди с СЭД имеют дефектный ген, который приводит к «слабому» коллагену или недостаточному количеству нормального коллагена. Это нарушает способность соединительной ткани поддерживать мышцы и органы.
В зависимости от типа СЭД, дефектный ген может быть унаследован от одного или обоих родителей. Иногда он не наследуется, а проявляется у человека впервые.
По данным 2016–2017 годов, как минимум у 1 человека из 5 000 есть проявления СЭД, причем 80–90% проявлений — это подвижность суставов. По приблизительным оценкам, около 2 млн человек в Великобритании, 10 млн в США, 17 млн в Европе и 255 млн во всем мире могут иметь такой синдром. Некоторые из редких, тяжелых проявлений СЭД опасны для жизни.
Чтобы поддержать больных с такими формами синдрома, в 1985 году была основана международная некоммерческая организация «Общество Элерса — Данлоса» (Ehlers-Danlos Society). Общество помогает проводить научные исследования СЭД и работает над распространением информации о синдроме. Логотип Общества — зебра, произошел от известного на Западе выражения: «Когда слышишь стук копыт, думай, что это лошади, а не зебры». Подразумевается, что при постановке диагноза врачи часто не принимают во внимание необычные и малораспространенные заболевания. Нередки случаи, когда пациенту с СЭД ставят другие диагнозы, так как медицинские работники не осведомлены о признаках синдрома.
История открытия синдрома Элерса — Данлоса
Еще в 400 году до н.э., Гиппократ заметил, что у кочевников и скифов вокруг суставов имеются следы прижиганий. Он посчитал, что этими шрамами и рубцами кочевники хотят как-то зафиксировать слишком подвижные суставы. В 1657 году голландский врач Иов Янсзон ван Микерин описал юношу 23 лет с настолько эластичной кожей, что он мог растянуть ее от подбородка вниз до груди и от колена до середины голени. При этом такая эластичность была только у правой половины его тела.
В 1892 году врач Мясницкой больницы в Москве Андрей Черногубов представил первое подробное описание синдрома. Он рассказал о двух пациентов с повышенной мобильностью крупных суставов. Один из них был 17-летний молодой человек с эпилепсией, у которого была «хрупкость и гиперэластичность кожи, неспособность удерживать швы, гипермобильность и вывих суставов, а также псевдоопухоли коленей, локтей и других областей». В России долгое время синдром Элерса — Данлоса (СЭД) был известен как синдром Черногубова.
Позднее, в 1901 году, датский дерматолог Эдвард Лауриц Элерс опубликовал описание болезни пациента со слабыми суставами и гиперэластичностью кожи, с предрасположенностью к образованию синяков. Семь лет спустя французский врач Анри-Александр Данлос (Данло) проводил осмотр пациента с похожими симптомами. В 1936 году английский дерматолог Фредерик Паркер Вебер объединил все случаи с гиперэластичностью и хрупкостью кожи, а также подвижностью суставов под общим названием «синдром Элерса-Данлоса».
Как проявляется синдром Элерса — Данлоса
Как отмечает доктор медицинских наук, профессор, директор Института методологии профессионального развития Анатолий Стремоухов, классификаций СЭД много, но самая удобная для понимания синдрома — классификация по клиническим проявлениям заболевания. Согласно ей, выделяют 10 типов СЭД. Все они имеют свои наборы признаков, некоторые типы схожи между собой.
Например, тип I — классический, «тяжелая форма», диагностируется чаще, чем другие. При СЭД первого типа у человека очень эластичная растяжимая кожа, которая легко подвергается ранам, кровотечениям. Шрамы образуются быстро, а заживают медленно. У человека с первым типом СЭД очень подвижные суставы, возможен деформированный скелет. Для женщины велика опасность преждевременных родов.
Второй, «мягкий», тип похож на классический, но имеет менее выраженные проявления: нет такой хрупкости и эластичности, как в первом случае, а подвижность наблюдается только в области стоп и кистей.
Третий тип СЭД — «гипермобильный», когда эластичность кожи минимальна, а вот суставы гиперподвижны. Около 90% всех случаев СЭД приходится на гипермобильный тип.
Есть и другие, менее распространенные типы. Клиника Мэйо, один из крупнейших частных медицинских и исследовательских центров мира, выделяет признаки и симптомы СЭД, характерные для большинства типов:
- Чрезмерная подвижность суставов. Боль в суставах и вывихи — обычное явление.
- Эластичная и легко растягивающаяся кожа, которая к тому же может быть необычайно мягкой и бархатистой.
- Хрупкая легко повреждаемая кожа, которая плохо заживает, на ней остается много тонких шрамов.
Тяжесть этих проявлений меняется от человека к человеку и зависит от конкретного типа синдрома.
Как врачи выявляют синдром Элерса — Данлоса
Кливлендская клиника, крупный частный медицинский центр США, объясняет, как врачи определяют СЭД у пациентов:
- Генетическое тестирование как самый распространенный способ выявления дефектного гена.
- Биопсия — тест, во время которого врач берет образец кожи и рассматривает его под микроскопом в поисках признаков заболевания, таких как специфические гены и генные мутации (аномалии).
- Физический осмотр, при котором врач определяет, насколько сильно растягивается кожа и насколько подвижны суставы.
- Рентгеновские снимки и компьютерная томография позволяют получить изображения внутренних частей тела, на которых можно увидеть отклонения от нормы, проблемы с работой сердца, искривление костей.
В России диагноз СЭД ставят на основе визуального осмотра, а также с помощью ЭМГ — электромиографии, которая позволяет оценить состояние скелетных мышц и нервов. Для определения типа СЭД можно сделать анализы на типы коллагена: многие частные клиники предлагают анализы на выявление синдрома.
Как синдром Элерса — Данлоса влияет на жизнь
Синдром по-разному влияет на человека в течение жизни. У него могут быть диагностированы другие заболевания: неврологические, связанные с позвоночником или сердечно-сосудистой системой. Например, сосудистый тип синдрома может привести к разрыву кровеносных сосудов, что, в свою очередь, может вызвать внутреннее кровотечение или инсульт. У людей с этим типом также повышен риск разрыва органов.
- боль в суставах и мышцах, в определенные периоды довольно сильная;
- слабость соединительных тканей, что может приводит к образованию грыж;
- изнуряющая хроническая усталость, которая негативно влияет на умственные и физические способности;
- заболевания сердца и сосудов;
- расстройства желудочно-кишечного тракта;
- гинекологические проблемы.
Кроме того, человек с СЭД может чувствовать головокружение, ухудшение памяти и внимания, изменения в потоотделении, иметь психологические трудности.
Лечение синдрома Элерса — Данлоса
Синдром невозможно вылечить, но существует поддерживающая терапия, смягчающая проявления СЭД. Общество Элерса — Данлоса выделяет ключевые идеи такой терапии:
- Подход к проводимому лечению должен быть целостным, сфокусированным на осложнениях и желании пациента уменьшить те проявления, которые мешают ему полноценно жить.
- Не следует ждать мгновенных результатов: часто требуются месяцы регулярных упражнений, чтобы остановить ухудшение состояния. Могут пройти годы, прежде чем пациент почувствует уменьшение боли. Хроническую усталость, как и боль, можно уменьшить с помощью лечебной физкультуры, но происходит это очень медленно.
- Комплексное применение лекарств, физиотерапии и дополнительных процедур работает гораздо лучше, чем редкий прием одного или двух лекарств. Некоторым пациентам может требоваться продуманная и широкая программа по избавлению от хронической боли.
- Общая цель терапии — научиться держать боль под контролем, сделать ее терпимой, так как устранить полностью ее, скорее всего, не получится.
- Когнитивно-поведенческая терапия и психологическое консультирование должны быть частью терапии СЭД.
В России пациенты получают поддерживающую терапию в зависимости от типа синдрома. Неврологические и сосудистые препараты может выписать генетик, также могут быть прописаны физиопроцедуры, ЛФК, курс массажа. Что касается получения инвалидности по СЭД, то с этим непросто: чаще можно получить инвалидность по другому тяжелому сопутствующему СЭД заболеванию. По утверждению самих больных, инвалидность по СЭД все же можно получить, если из-за синдрома происходит ограничение жизнедеятельности.
Известные люди с синдромом Элерса — Данлоса
В прошлом людей с СЭД можно было увидеть в цирках и странствующих шоу. Их называли «Эластичная леди» или «Резиновый человек» — из-за их способностей растягивать свою кожу и удивительной гибкости. В 1880 была сделана первая фотография человека с СЭД: это был Феликс Верле, акробат с эластичной кожей, который, помимо прочего, мог выгибать ноги в обратную сторону. Еще об одном «резиновом человеке», Джеймсе Моррисе, даже сняли в 1902 году документальный фильм.

От синдрома страдали и некоторые известные люди — например, у скрипача-виртуоза Никколо Паганини были очень подвижные суставы и деформация грудной клетки, что, возможно, указывает на то, что у него тоже был СЭД.
Среди людей с синдромом есть артисты, модели, спортсмены. Американская певица Сиа поделилась, что живет с синдромом и страдает от хронической боли. А певица Мика Дельгадо, солистка поп-панк группы Yours Truly, даже записала песню о своем опыте жизни с СЭД.

Аллиза Сили — американская паратриатлонистка. После того, как в 2010 году у нее диагностировали синдром Элерса — Данлоса с сопутствующими заболеваниями, Сили прошла через ряд серьезных медицинских вмешательств, включая ампутацию левой ноги. Это не помешало ей завоевать золотую медаль в женском триатлоне на летних Паралимпийских играх 2016 года.
Синдром Эллерса-Данло тип VI, PLOD ч.м.
Синдром Элерса - Данло тип VI (EDS, СЭД, OMIM225400) - гетерогенная группа наследственных заболеваний соединительной ткани, в основе которых лежит недостаточное развитие коллагеновых структур в различных системах организма. Назван в честь двух дерматологов, Эварда Элерса и Генри Данлоса. Но ещё раньше синдром был описан Николаем Черногубовым. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз. Относится к моногенным заболеваниям с различными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена, одного из основных белков соединительной ткани. Конечный результат каждого из механизмов формирования патологии один и тот же - уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса - Данло обусловлена тем, что элементы поражённой соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Согласно классификации наследственных болезней соединительной ткани, выделяют 9 форм синдрома Элерса - Данло, различающихся особенностями клинической картины и типом наследования. Характерными признаками синдрома Элерса - Данло типа VI являются аутосмно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена — одного из основных белков соединительной ткани, приводящих к уменьшение стабильности коллагенового волокна. Элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма, что приводит к генерализованному поражении организма при заболевании. Характерными признаками синдрома Элерса-Данло типа VI являются: мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
- Мутация не выявлена.
- Мутация выявлена в гетерозиготном состоянии.
- Мутация выявлена в гомозиготном состоянии.
- Мутация выявлена в компаунд –гетерозиготном состоянии.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации. ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Сосудистый тип синдрома Элерса–Данло – редкое моногенное заболевание соединительной ткани
Синдром Элерса–Данло – редкое (орфанное) заболевание, характеризующееся дисплазией соединительной ткани, хрупкостью кровеносных сосудов и тканей и вариабельной клинической картиной. Сосудистый тип синдрома Элерса–Данло, относящийся к группе А, согласно классификации 2017 г. обусловлен мутациями в гене альфа-1-цепи коллагена III типа COL3A1. Заболевание отличается высокой летальностью пациентов вследствие спонтанных разрывов стенок сосудов и полых внутренних органов. Международным Консорциумом (2017) разработаны критерии клинической диагностики сосудистого типа синдрома Элерса–Данло.
Представлено клиническое наблюдение пациента 16 лет мужского пола с синдромом Элерса–Данло сосудистого типа. При молекулярно-генетическом исследовании у ребенка выявлена ранее описанная патогенная мутация сайта-сплайсинга p.Gly798_Pro815del гена COL3A1, связанная с тяжелым течением заболевания. Несмотря на комплекс лечебных мероприятий, направленных на укрепление сосудистой стенки, стимуляцию и нормализацию энергетического и минерального обменов, через 10 мес наступил летальный исход вследствие разрыва аорты и почечной артерии. Приведено заключение судебно-медицинской экспертизы. Результаты представленного наблюдения свидетельствуют, что во избежание пропуска больных целесообразно пересмотреть минимальный набор признаков, необходимых для установления клинического диагноза.
Ключевые слова
Об авторах
ОСП «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
Россия
Семячкина Алла Николаевна – д.м.н., гл. науч. сотр. отдела клинической генетики
125412 Москва, ул. Талдомская, д. 2
ОСП «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
Россия
Николаева Екатерина Александровна – д.м.н., рук. отдела клинической генетики
125412 Москва, ул. Талдомская, д. 2
ОСП «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
Россия
Данцев Илья Сергеевич – врач-генетик педиатрического отделения врожденных и наследственных заболеваний
125412 Москва, ул. Талдомская, д. 2
ОСП «Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
Россия
Меликян Люся Петросовна – науч. сотр. отдела клинической генетики
125412 Москва, ул. Талдомская, д. 2
Танатологическое отделение №1 Бюро судебно-медицинской экспертизы Департамента здравоохранения города Москвы
Россия
Павлова Мария Сергеевна – судебно-медицинский эксперт
115516 Москва, Тарный проезд, д. 3, стр. 2
Список литературы
1. Malfait F., Francomano C., Byers P., Belmont J., Berglund B., Black J. et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet C Semin Med Genet 2017; 175(1): 8–26. DOI: 10.1002/ajmg.c.31552
2. Germain D.P. Ehlers–Danlos syndrome type IV. Orphanet J Rare Dis 2007; 2:32. DOI: 10.1186/1750-1172-2-32
3. Eagleton M.J. Arterial complications of vascular Ehlers–Danlos syndrome. J Vasc Surg 2016; 64(6): 1869–1880. DOI: 10.1016/j.jvs.2016.06.120
4. Papagiannis J. Sudden death due to aortic pathology. Cardiol Young 2017; 27(S1): S36–S42. DOI: 10.1017/S1047951116002213
6. Shields L.B.E., Rolf C.M., Davis G.J., Hunsaker J.C. Sudden and unexpected death in three cases of Ehlers–Danlos syndrome type IV. Case Reports. J Forensic Sci 2010; 55(6): 1641–5. DOI: 10.1111/j.1556-4029.2010.01521.x
7. Park M.A., Shin S.Y., Kim Y.J., Park M.J., Lee S.H. Vascular Ehlers–Danlos syndrome with cryptorchidism, recurrent pneumothorax, and pulmonary capillary hemangiomatosis-like foci: A case report. Medicine (Baltimore) 2017; 96(47): e8853. DOI: 10.1097/MD.0000000000008853
8. Park K.Y., Gill K.G., Kohler J.E. Intestinal Perforation in Children as an Important Differential Diagnosis of Vascular Ehlers–Danlos Syndrome. Am J Case Rep 2019; 20: 1057–1062. DOI: 10.12659/AJCR.917245
9. D’hondt S., Van Damme T., Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers–Danlos syndrome: a systematic review. Genet Med 2018; 20(6): 562–573. DOI: 10.1038/gim.2017.138
Рецензия
Для цитирования:
For citation:
Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Резиновый человек
Синдром Э́лерса — Данлóса (по некоторым источникам синдром Э́лерса — Данлó, СЭД) — это редкая наследственная патология коллагена, поражающая кожу, опорно-двигательный аппарат и другие органы. Другие названия синдрома — «гиперэластичность кожи», сutis hyperelastica, несовершенный десмогенез Русакова и синдром Черногубова — Элерса — Данлоса. Людей с этим синдромом в прошлом можно было увидеть в цирке из‑за их способности удивлять обывателей своей внешностью и движениями. Этой болезнью страдали и некоторые известные люди — например, скрипач-виртуоз Никколо Паганини.
История открытия синдрома
Людей с этой болезнью описывал еще Гиппократ в 400 году до н. э. в сочинении «Воздухи, воды и места» (De aere, aquis, locis). Он наблюдал за кочевниками и скифами со слабыми суставами и множественными шрамами около них. Гиппократ посчитал, что шрамы были следами прижигания, при помощи которого пытались снизить гипермобильность суставов. Много позже — в 1657 году — голландский хирург Джансун ван Микерен (J. Van Meekeren, 1611–1666 гг.) описал маленького испанца с очень эластичной кожей. Мальчик по имени Джордж Альбс мог растянуть кожу подбородка до груди, а кожу на коленях до середины голени, однако это касалось лишь правой половины его тела. Всемирно известный скрипач и композитор Никколо Паганини (1782–1840 гг) имел гипермобильные суставы, тонкое телосложение и деформацию грудной клетки — все симптомы, характерные для носителя синдрома Элерса — Данлоса. В конце XIX века некоторые пациенты с СЭД выступали в качестве людей с необычными способностями в передвижных шоу — например, «эластичная леди», описанная американскими врачами Джорджем Гулдом и Вальтером Пайлом (George M. Gould, Walter L. Pyle). Разнообразие клинических проявлений синдрома долгое время не позволяло подробно описать все формы заболевания.
Классификация и название
Первое подробное описание СЭД представил русский врач Мясницкой больницы в Москве Андрей Черногубов на Московском венерологическом и дерматологическом обществе в 1892 году. Он описал двух пациентов с повышенной мобильностью крупных суставов. Один из них — 17‑летний парень с эпилепсией, обладавший хрупкой и гиперэластичной кожей, неспособной удерживать швы.
Позднее, в 1901 году, датский дерматолог Эдвард Лауриц Элерс (Edvard Lauritz Ehlers) опубликовал описание пациента со слабыми суставами и гиперэластичностью кожи, с предрасположенностью к образованию синяков. Его же он продемонстрировал на Дерматологическом обществе Дании в 1899 году. Семь лет спустя французский врач Анри-Александр Данлос(Данло) (Henri-Alexandre Danlos) осмотрел пациента с сосудистым поражением кожи на локтях и коленях. После появлялись отдельные описания этого синдрома и в США, и в Англии, и к 1966 году общее число докладов возросло до 300. В 1936 году английский дерматолог Фредерик Паркер Вебер (Frederick Parkes Weber) объединил все случаи с гиперэластичностью и хрупкостью кожи, а также гипермобильностью суставов. Он назвал новое заболевание «синдромом Элерса — Данлоса».
В 1972 году был обнаружен первый молекулярный дефект коллагена при СЭД. В 1986 году на Международном конгрессе по наследственным заболеваниям соединительной ткани в Берлине было выделено 9 типов синдрома Элерса — Данлоса, но в 1997 году в городе Вильфранш-сюр-Мер (Франция) эксперты разработали и приняли более точную классификацию. В ней было уже 6 типов СЭД:
- классический;
- гипермобильный;
- сосудистый;
- кифосколиотический;
- ахондроплазипластический;
- дерматоспараксис.
Критерии диагноза
Эта классификация не только систематизирует формы синдрома, но и выделяет большие и малые критерии диагностики синдрома Элерса — Данлоса, поэтому ее часто называют Вильфраншскими диагностическими критериями.
Большие диагностические критерии:
- Тонкая просвечивающая кожа с проступающим венозным рисунком.
- Предрасположенность к сосудистым, кишечным и маточным разрывам или слабости этих структур.
- Легкое образование синяков и кровоточивость.
- Характерные черты лица: широко посаженные глаза, вдавленная средняя часть и эпикантус складка у внутреннего угла глаза, прикрывающая слёзный бугорок
Некоторые малые диагностические критерии:
- Преждевременное старение конечностей (акрогерия — атрофия кожи кистей и стоп).
- Гипермобильность малых суставов (межфаланговых и пястно-фаланговых суставов кисти).
- Разрыв сухожилий и мышц.
- Косолапость.
- Пневмоторакс/пневмогемоторакс.
- Ретракция (оседание) десен, их недоразвитие.
- Отягощенный семейный анамнез, внезапная смерть близких родственников.
Два или более больших критерия позволяют заподозрить СЭД, для уточнения требуется лабораторное подтверждение. Если имеют место только малые критерии, то это не СЭД — должен быть хотя бы один большой критерий.
Патогенез и клиника
СЭД — это следствие мутаций в различных генах. Мутации затрагивают гены, преимущественно задействованные в синтезе коллагена, в результате чего его волокна имеют неправильную форму и ориентацию. Они располагаются беспорядочно, что и приводит к основным клиническим проявлениям синдрома Элерса — Данлоса. Мутации могут возникать спорадически, но известны и семейные случаи. Типы СЭД выделены на основании анализа доминирующего симптомокомплекса.
Гипермобильный СЭД
Наследуется по аутосомно-доминантному типу и связан с геном коллагена 3 типа COL3A1. Встречается у одного человека на 10 000–15 000 населения. Доминирующий симптом — повышенная мобильность крупных и мелких суставов, сопровождающаяся болью. Вывихи и подвывихи могут возникать спонтанно или из‑за незначительных травм. Симптомы возникают вне зависимости от возраста, однако у детей с синдромом Элерса — Данлоса поставить диагноз труднее вследствие физиологической слабости связочного аппарата суставов. Пациентам с этим типом СЭД также свойственно раннее развитие остеопороза (практически сразу после 30 лет). Для этого вида СЭД характерны так же функциональные расстройства толстого кишечника, гипермобильность пищевода, гастроэзофагеальный рефлюкс и гастрит.
Классический СЭД
Связан с дефектом коллагена V и I типа вследствие мутации в гене COL5A1 и других генах семейства коллагенов. Встречается у одного на 20 000–40 000 человек. Доминирующее проявление — повышенная растяжимость кожи, которая так же сопровождается геморрагическим синдромом, шрамы и раны заживают нетипичным образом, нередко формируются кисты под кожей, возникают доброкачественные новообразования кожи и подкожной клетчатки.
Сосудистый СЭД
Развивается при аутосомно-доминантном дефекте гена COL3A1, участвующего в синтезе коллагена типа III. Встречается реже, чем предыдущие два типа — у одного на 250 000 человек. Этот тип характеризуется высоким риском профузных кровотечений из внутренних органов и разрывов кровеносных сосудов. Пациент с сосудистым типом СЭД обладает тонкой кожей с просвечивающими сквозь нее сосудами. Именно из‑за проблем с сосудами такие больные редко доживают до 50 лет. Внешний вид пациента с сосудистым типом СЭД может быть весьма характерным — лицо выглядит истощенным с выступающими скулами и впалыми щеками, нос и губы тонкие. При этом выраженной гиперэластичности кожи нет.
Лечение и прогноз
Пациенты с СЭД в течение жизни наблюдаются разными специалистами — терапевтами, генетиками, ортопедами, физиотерапевтами, специалистами ЛФК, неврологами, кардиологами и др., в зависимости от клинических симптомов. Специфического лечения не существует. Ранняя диагностика синдрома Элерса — Данлоса у детей позволяет составить прогноз, подобрать необходимый образ жизни пациенту и снизить количество осложнений, связанных с основным заболеванием. Большинство людей с этим диагнозом могут прожить относительно нормальную и долгую жизнь. Главное условие — снизить травматизм при сохранении достаточной физической нагрузки для развития мышечного каркаса. Кроме того, необходимы регулярные профилактические осмотры и лечение у стоматолога и офтальмолога.
Читайте также: